Improving outcomes in advanced melanoma with systemic therapy
Melanoma is extremely challenging to treat in the later stages. Two new agents – the monoclonal antibody ipilimumab and the BRAF inhibitor vemurafenib – have received both FDA and EMA approval for use in advanced (unresectable or metastatic) melanoma.
Dr Giuseppe Gullo, Consultant Medical Oncologist, St Vincents University Hospital, Dublin
Melanoma is the fourth most commonly diagnosed cancer in women and the fifth in men in Ireland. The most recent figures from the Irish National Cancer Registry indicate that with more than 700 new cases per year, melanoma represents 4.1% of all invasive cancers in both sexes.
It is well known that the incidence of melanoma is increasing faster than other types of cancer. In fact, the annual percentage change in melanoma incidence rate in Ireland between 1994 and 2009 is statistically significant, with a 2.1% and a 4.8% increase in women and men, respectively.1
Melanoma is highly curable when diagnosed in the earlier stages but is extremely challenging to treat in the later stages because of the lack of response to virtually all the anticancer therapeutics.
The only drugs approved in Europe and the US for treatment of advanced melanoma prior to 2011 were high-dose interleukin-2 and dacarbazine (DTIC), which both have response rates of only 10-20%.2,3 Until recently, none of the agents used to treat melanoma demonstrated an improvement in overall survival, thus emphasising the need for novel agents.
The scenario of systemic therapy for advanced melanoma changed dramatically in 2010 when, for the first time ever, a single drug was found to prolong survival of patients pre-treated with systemic therapy.
As of January 2012, only 18 months later, two new agents – the monoclonal antibody ipilimumab and the BRAF inhibitor vemurafenib – have received both Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval for use in advanced (unresectable or metastatic) melanoma, and there is an exciting flourishing of pre-clinical and clinical research in the field. Such a major change in a remarkably short period of time is unique in the treatment of a human malignancy.
The most recent and significant advancements in melanoma treatment involve:
The modulation of the interactions between the host immune system and the cancer cells
Targeting the molecular pathways involved in the uncontrolled proliferation of melanoma cells.
Ipilimumab
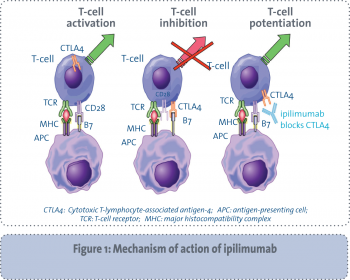
Ipilimumab is a fully humanised monoclonal antibody directed at the cytotoxic T lymphocyte-associated antigen 4 (CTLA4). This is a T-cell surface receptor that works as an immune system checkpoint to regulate immune responses. CTLA4 is expressed when the immune system is stimulated and competes with CD28 for binding on antigen presenting cells, thus leading to blockade of costimulatory signals needed for T-cell activation.
As shown in Figure 1, blockade of CTLA4 releases immune system inhibition allowing for enhanced T-cell-mediated immunity and the ability to recognise cancer cells as foreign.4
(click to enlarge)
Pre-clinical data showed ipilimumab to be effective in enhancing the host’s ability to generate an anticancer immune response by depletion of regulatory T-cells. Ipilimumab can also be successfully combined with cytotoxic agents in order to increase anti-tumour activity on melanoma cell lines.
Following the encouraging results of several smaller phase II clinical studies, ipilimumab took the clinical oncology world by storm when the results of a large phase III trial were presented at the American Society of Clinical Oncology (ASCO) Annual Meeting in Chicago in June 2010.
In this trial, 676 patients with pre-treated, unresectable stage III or stage IV melanoma were randomised to receive ipilimumab at 3mg/kg intravenously every three weeks, with or without a glycoprotein 100 (gp100) vaccine versus the gp100 vaccine alone.
Patients treated with ipilimumab and gp100 had an overall survival of 10 months compared with 6.4 months in the gp100 control group (p < 0.001).
The best overall response rate was 10.9% and disease control rate including complete response, partial response and stable disease was 28.5% in patients treated with ipilimumab. There was no difference in progression-free survival (PFS) in patients treated with ipilimumab and gp100 compared with gp100 alone (2.76 months), which highlights the latency period of about three months for the drug to take effect. The one- and two-year survival rates for patients treated with ipilimumab and gp100 compared with gp100 alone were 45.6% and 23.5%, respectively.5
At the ASCO Annual Meeting in 2011, Dr Jedd Wolchok, director of Immunotherapy Clinical Trials at New York’s Memorial Sloan-Kettering Cancer Center, presented the results of a phase III study of dacarbazine (DTIC) and ipilimumab versus DTIC and placebo in patients with previously untreated advanced melanoma. The results were subsequently published in the New England Journal of Medicine.6
In this study, 502 treatment-naïve patients with metastatic melanoma and ECOG (Eastern Cooperative Oncology Group) performance status 0/1 were randomised 1:1 to ipilimumab (10mg/kg) plus DTIC (850mg/m2) or placebo and DTIC (850mg/m2) at weeks one, four, seven and 10 followed by DTIC every three weeks through week 22 (induction). Eligible patients received ipilimumab or placebo every 12 weeks as maintenance.
Overall survival rates for combination therapy versus chemotherapy alone after one, two and three years of therapy were: 47.3% versus 36.3%, 28.5% versus 17.9%, and 20.8% versus 12.2%, respectively.
Median overall survival was 11.2 months with ipilimumab plus DTIC versus 9.1 months for DTIC alone. Median progression-free survival (PFS) times, however, were similar across the two arms with 2.8 months for combination therapy versus 2.6 months for DTIC alone.The lack of difference in PFS confirms the findings from the second-line trial that the effects of immunotherapy can take much longer to be seen than those from traditional chemotherapy or targeted therapies. In view of this, overall survival is likely to be a more accurate way to gauge treatment effectiveness than PFS when immunomodulating agents are used.
Given its unique mechanism of action, ipilimumab has a toxicity profile that differs from the vast majority of other anti-cancer therapeutics currently in clinical use. This requires a careful monitoring of patients undergoing treatment with ipilimumab and a specific education of the treating physicians. The two phase III trials presented above5,6 showed slightly different toxicity profiles, most likely reflecting the differences in the study populations and in the agents that were given in combination with ipilimumab (gp100 vaccine and DTIC, respectively).
Immune-related adverse events (IRAEs), including dermatitis, hepatitis, nephritis, hypophysitis and, more commonly, diarrhoea and enterocolitis, are seen with ipilimumab. In the pre-treated trial of ipilimumab, 60% of patients experienced an IRAE, with 10-15% being grade 3 or 4.
Most of these adverse events remit within six weeks of discontinuing ipilimumab. In the ipilimumab/gp100 trial, there were 14 deaths related to the study drugs (2.1%), and seven were associated with IRAEs.5
Diarrhoea and enterocolitis are two common IRAEs, occurring in about 20-30% of treated patients, although grade 4 colitis is rare. Prompt initiation of treatment, including steroids, is effective in alleviating symptoms in the vast majority of patients within two weeks, although a small proportion of patients require treatment with infliximab, an antibody against tumour necrosis factor (TNF-α ).7 Severe enterocolitis leading to colonic perforation and subsequent death has been reported in the literature.
In the first-line trial, the combination of ipilimumab and DTIC had a good safety profile, with no gastrointestinal perforations and a lower rate of colitis than in the prior ipilimumab monotherapy studies. However, approximately 56% of patients receiving ipilimumab plus DTIC and 27% of patients receiving DTIC alone had significant grade 3 or 4 adverse events from their therapy.6
Ipilimumab is the first agent ever proven to improve survival in advanced melanoma. The positive results of the first-line phase III trial substantially confirmed the magnitude of benefit of ipilimumab seen in the previous trial on advanced melanoma patients, and led to the approval in 2011 of ipilimumab for patients with advanced melanoma in first- and second-line treatment by the FDA and in second-line treatment by the EMA.
Vemurafenib
BRAF, a serine/threonine kinase member of the mitogen-activated protein kinase (MAPK) pathway, has been found to have an activating mutation in approximately 50% of melanomas.8 Over 90% of the mutations in BRAF are due to a glutamic acid (E) for valine (V) amino acid substitution at position 600 (V600E), which causes an unregulated signalling of the MAPK pathway and thus uncontrolled cell growth.
Preclinical studies on human melanoma cell lines have shown that inhibition of BRAF by small inhibiting RNAs is able to restore cell turnover arrest and thus reverse the malignant features of the melanoma cell lines (see Figure 2).9 Therefore BRAF inhibition appears an attractive and potentially successful therapeutic strategy for the sub-group of patients with an activating mutation of BRAF.
(click to enlarge)
Vemurafenib is a small molecule with inhibitory activity that targets the mutant BRAF at the V600E mutation.10 Following the observation of a significant anti-tumour activity of this agent on patients with BRAF-mutated melanoma in a phase I dose-escalation trial, vemurafenib was further investigated in a phase II study (BRIM-2) conducted on this molecularly-defined sub-population of melanoma patients. In this trial 52% of patients (all pre-treated with at least one line of systemic therapy) having a partial or complete response and 29.5% stable disease. At a median follow-up of seven months, the duration of response was 6.8 months and PFS was 6.2 months. Around 69% of patients were still alive at the time of data analysis and median overall survival had not yet been reached.11
The results of the BRIM-3 trial were presented at the 2011 ASCO Annual Meeting and led to the FDA approval of vemurafenib in August 2011 as first-line therapy for patients with BRAF V600E mutation-positive advanced melanoma.12
On December 15, 2011, the EMA’s Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for vemurafenib for the treatment of patients suffering from metastatic or unresectable melanoma with BRAF V600 mutations. At the time of going to press, this recommendation is with the European Commission for the adoption of a binding decision.
The BRIM-3 study enrolled 675 treatment-naïve patients with stage IIIc or IV BRAF V600E mutant melanoma and compared the effectiveness (overall survival and progression free survival as co-primary endpoints) of treatment with vemurafenib (960mg orally twice daily) or DTIC (1,000mg/m2 intravenously every three weeks) as first-line therapy.
At the planned interim analysis, patients receiving vemurafenib had a 63% reduction in risk of death [95% CI 0.26-0.55; p < 0.0001] compared with those receiving DTIC. There was also a 74% reduction in the risk of progression (or death) with vemurafenib compared with DTIC (95% CI 0.26-0.55; p < 0.0001). Response rates were 48.4% with vemurafenib versus 5.5% with DTIC. Due to this result, it was recommended that patients receiving DTIC crossover to receive vemurafenib.
Vemurafenib seems to be generally well tolerated with adverse events including grade 2 or 3 arthralgias, rash, nausea, pruritus, photosensitivity, palmar-plantar dysesthesia and fatigue. Cutaneous reactions including keratoacanthoma-type squamous cell carcinomas were seen in about 20-30% of patients.
This seems to be a class-specific adverse event of BRAF inhibitors but as these are slow-growing neoplasms, with little risk of significant local invasion or distant metastases; complete surgical excision is curative. Less than 10% of patients who received vemurafenib in the BRIM-3 trial experienced problems with high levels of toxicity (grade 3 or worse).
It has been found that some patients appear to have primary resistance to vemurafenib whereas another group may develop secondary resistance as they become resistant after initially responding to the drug.11
Experiments in melanoma cell lines indicate that those cells that became resistant to the BRAF inhibitor vemurafenib can be completely destroyed by the combination of BRAF and MEK inhibition.13 These data provide support for simultaneous use of BRAF and MEK inhibitors to target the population of primary resistant cells. Several ongoing clinical trials using the combination of BRAF and MEK targeted agents will be able to test this hypothesis.
Ipilimumab and vemurafenib in clinical practice
Ipilimumab has been reviewed and approved by the EMA and the FDA, and vemurafenib is anticipated to receive European Commission marketing approval in the near future. Many medical oncologists in Ireland have already had the opportunity to use both these two agents to treat their patients in the context of clinical trials and/or with the ongoing expanded access programmes.
Nevertheless, the optimal use of ipilimumab and vemurafenib in clinical practice is not yet clear and their use should be based on the characteristics of each drug and on each patient’s tumour characteristics.
Ipilimumab and vemurafenib are significantly different in terms of mechanisms of action and patient subgroups in which they have been investigated. The two phase III trials of ipilimumab5,6 consistently show that only a minority of patients, estimated to be between 20-30% of the total, will eventually benefit from the CTLA4 inhibition. These patients may achieve a long-term disease control that in some cases may last for years.
Unfortunately, no biomarkers have been identified to date that allow the selection in advance of these ipilimumab-sensitive patients. At present, approximately three quarters of patients are treated with ipilimumab without seeing any treatment benefit while being exposed to the risk of harmful side effects.
Vemurafenib, however, has been clinically developed in a molecularly defined subgroup and the presence of the V600E mutation is an excellent predictor of vemurafenib activity. Unfortunately, despite the extremely high response rate and the prolonged progression-free and overall survival, mechanisms of resistance to the BRAF inhibition have been found to develop after a certain period of time in the majority of responding patients and there is a low probability of long-term responses.11,12
It should be also noted that the median follow up in the vemurafenib trials is significantly shorter than in the ipilimumab trials thus not enabling, at the moment, head-to-head comparisons of the long-term activity of these two agents.
Another major difference between ipilimumab and vemurafenib is the time to response in treated patients. As ipilimumab produces its effects only through the elicitation of an immune response against the melanoma cells in the host, there is an expected latency period prior to observing any significant objective response.
During this time, which typically covers the 12-week induction phase, some patients’ scans may actually get worse before they improve.
This is a challenge to the current paradigm in assessing tumour response to systemic therapies and must be taken into consideration by the treating oncologist before making any decision on discontinuation of ipilimumab during the induction phase.
In the present scenario, all patients with unresectable stage III or stage IV melanoma should be tested upfront for the presence of BRAF mutations. Patients with BRAF V600E mutation-positive melanomas should receive a BRAF inhibitor, such as vemurafenib or any another similar investigational agent in a clinical trial.
Although there is no data on the activity of ipilimumab in the vemurafenib pre-treated patients, in view of the different mechanism of action of these two agents, it is reasonable to assume that ipilimumab would retain its activity in this setting of patients.
Similarly, there is no data at present on the activity of ipilimumab in patients with mutated BRAF. Given the possibility of long-term disease control in a small subgroup of advanced melanoma patients,
it is acceptable to consider a first-line treatment with ipilimumab in BRAF mutation-positive patients but only when the disease is not rapidly progressive and a time to response of three months is not likely to affect patient’s outcome.
Conclusion
The therapeutic scenario of advanced melanoma has changed dramatically in the last year thanks to the availability of ipilimumab and vemurafenib. We are only at the beginning of a new and revolutionary process that has brought translational research to melanoma care. It is now of paramount importance that all cancer professionals support the second phase of clinical trials, which are testing new agents and new combinations of agents in order to overcome mechanisms of resistance and improve outcome.
Ireland can play a major role in this research effort given the high level of oncology care and new melanoma referral guidelines in the country.
The management of metastatic melanoma poses a distinct challenge and requires ongoing laboratory and clinical research efforts but the recent development of these new agents has for the first time in over 30 years changed the survival outlook for patients and brought hope that significant progress can be achieved.
References
Cancer in Ireland in 2011: Annual Report of the National Cancer Registry. Available at http://www.ncri.ie/ncri/index.shtml Accessed Dec 18, 2011
Agarwala SS. Current systemic therapy for metastatic melanoma. Expert Rev Anticancer Ther 2009; 9: 587-595
Atkins MB, Lotze MT, Dutcher JP et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999; 17: 2105-2116
Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opion Immunol 2006; 18: 206-213
Hodi FS, O’Day SJ, McDermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. NEJM 2010; 363: 711-723
Robert C, Thomas L, Bondarenko I et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. NEJM 2011; 364: 2517-2526
Beck KE, Blansfield JA, Tran KQ et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol 2006; 24: 2283-2289
Davies H, Bignell GR, Cox C et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949-954
Hingorani SR, Jacobetz MA, Robertson GP et al. Suppression of BRAF (V599E) in human melanoma abrogates transformation. Cancer Res 2003; 63: 437-450
Tsai J, Lee JT, Zhang J et al. Discovery of a selective inhibitor of oncogenic BRAF kinase with potent antimelanoma activity. Proc Natl Acad Sci USA 2008; 105: 3041-3046
Flaherty KT, Puzanov I, Kim KB et al. Inhibition of mutated, activated BRAF in metastatic melanoma. NEJM 2010; 363: 809-819.
Chapman PB, Hauschild A, Robert C et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. NEJM 2011; 364: 2507-2516
Paraiso KHT, Fedorenko IV, Cantini LP et al. Recovery of phosphor-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer 2010; 102: 1724-1730
 (click to enlarge)
(click to enlarge)
