Novel therapies have become the mainstay for MM. As our knowledge of disease pathophysiology expands so does the search for newer treatment modalities with fewer side-effects
Dr Sally Mohammed, Registrar in Haematology, Midland Regional Hospital, Tullamore, Co Offaly, Ms Mary B Kelly, Advanced Nurse Practitioner in Haematology, Midland Regional Hospital, Tullamore, Co Offaly, Dr Liam Smyth, Specialist Registrar in Haematology, Midland Regional Hospital, Tullamore, Co Offaly and Dr Gerard Crotty, Consultant Haematologist, Midland Regional Hospital, Tulllamore, Co Offaly
Multiple myeloma (MM) is a malignant plasma cell disorder that accounts for approximately 10% of all haematological cancers.1,2 It is now known that, in most cases, MM evolves from an asymptomatic premalignant stage of clonal plasma cell proliferation termed ‘monoclonal gammopathy of undetermined significance’ (MGUS). MGUS is present in more than 3% of the population above the age of 50 and progresses to MM or related malignancy at a rate of 1% per year.
MM has traditionally been considered a disease of the older population but younger patients are increasingly being diagnosed. In fact, 5% of patients are aged less than 50 years old and 2% less than 40.
In the Republic of Ireland, the most recently reported incidence data state that 207 new patients are diagnosed with MM on average every year.3 However, the prevalence, ie. the number of patients living with the disease, has increased, largely owing to the advances in disease management and control.
MM remains a disease without a cure and the presenting symptoms and signs – increased serum calcium, renal impairment, anaemia and bone lesions, or ‘CRAB’ – are unchanged from the time it was first recognised as a disease entity in the early 19th century. Although this holds true, astonishing advancements have been made in all aspects of MM diagnosis and care.
In this article we tell the story of MM, with specific reference to the evolution of diagnostic methods, treatment modalities and disease control measures and the subsequent lengthening of patient survival and improvement in quality of life.
Identification and description
MM has probably been present for thousands of years, however, the first well-documented case was a patient described by Solly in 1844.4 Sarah Newbury, a 39-year-old woman, developed fatigue and pain from multiple bone fractures.
At autopsy, four years after the onset of symptoms, the bone marrow was found to be replaced by a red substance whose cells were very similar to those found at the autopsy of Thomas Alexander McBean, another patient described.
Solly thought the disease was an inflammatory process. In his description he also alluded to a vascular process, and it has been sentimentalised that he may have been contemplating the angiogenic nature of the pathogenesis of the disease.4
McBean was a patient who was seen in 1845 by Dr William Macintyre in London, who examined his urine and sent the following note and a sample of urine to Henry Bence Jones, a physician and chemist.5
Saturday, November 1, 1845:
Dear Dr Jones, The tube contains urine of very high specific gravity. When boiled it becomes slightly opaque. On the addition of nitric acid, it effervesces, assumes a reddish hue, and becomes quite clear; but as it cools, assumes the consistence and appearance which you see. Heat liquefies it. What is it?
Henry Bence Jones studied the urine from Mr McBean in detail and confirmed the physical properties described by Macintyre, referring to the unusual thermo-solubility characteristics of the urine containing this protein that precipitates when heated to 40-60°Celsius and re-dissolves on boiling.6 Jones erroneously concluded that the protein was related to albumin but emphasised its role in the diagnosis of MM.
In current practice, the Bence Jones protein remains an important marker for the diagnosis and monitoring of MM. However, the way it is detected has evolved to far more sophisticated methods using the now-standard method of immune-fixation electrophoresis or, more recently, automated nephelometric assays for free kappa and lambda light chains that do not detect immunoglobulin-bound light chains.7
Identification of the serum monoclonal protein
Hyperproteinaemia was first demonstrated in MM in 1928 by Perlzweig et al.8 Seven years later, Tiselius9 separated serum globulins into three components, which he designated as alpha, beta and gamma. Immune-electrophoresis was described by Grabar and Williams in 1953.10 Eleven years later, immune-fixation was introduced into clinical practice by Wilson.11
An important development was the distinction between monoclonal and polyclonal gammopathies outlined by Jan Waldenström in 196112. He described a narrow band of hypergammaglobulinaemia on electrophoresis as being a monoclonal protein. Many of the patients with this feature had MM or macroglobulinaemia, but others had no evidence of malignancy. He considered them to have ‘essential hypergammaglobulinaemia’ or a ‘benign monoclonal protein’.
Robert Kyle favoured the term ‘monoclonal gammopathy of undetermined significance’ (MGUS), as this emphasised the need for follow-up and the potential for neoplastic evolution, whereas the word ‘benign’ would usually imply the lack of need for such follow-up.13
The ability to differentiate between monoclonal versus polyclonal gammopathies is important because patients with a polyclonal gammopathy have an inflammatory or reactive process, whereas those with a monoclonal gammopathy either have a malignancy or may develop a neoplastic process.14
Staging and risk stratification
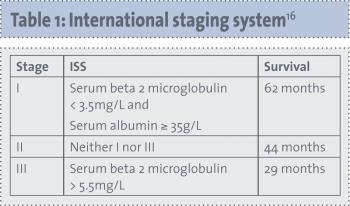
The Durie-Salmon staging system has been used to stratify patients with MM since 1975.15 This system is based on the CRAB criteria as well as the serum paraprotein level and urinary excretion of light chains. More recently, Greipp et al16 developed an international staging system, which requires knowledge of the patient’s beta-2 microglobulin and albumin levels. Table 1 shows the international staging system.
(click to enlarge)
Alongside stage, important prognostic factors that stratify patients into high risk and standard risk are cytogenetic abnormalities such as deletion of chromosome 13 or hypodiploidy on karyotyping, 17p deletion or immunoglobulin heavy chain translocations t(4;14) or t(14;16) on molecular genetic studies.17 The presence of any one or more of the high-risk factors classifies a patient as having high-risk MM. The median survival of patients with high-risk features is two to three years, compared with five or more years in patients without the high-risk factors.
Evolution of management
It is important to note that treatment is not always indicated in MM, particularly in the asymptomatic patient, but close monitoring is required. There is no evidence that early treatment of patients with asymptomatic (smouldering) MM prolongs survival. However, clinical trials are ongoing to determine whether newer agents can delay progression.
The treatment of MM has changed markedly from the days of rhubarb pills, infusion of orange peel, the use of phlebotomy and leeches. In 1947, urethane, also known as the chemical compound ethyl carbamate, was reported to produce a reduction in serum globulin, an increase in haemoglobin, disappearance of proteinuria, and a decrease in bone marrow plasma cells in a patient with MM. This was the standard of therapy for more than 15 years.18
In 1958, Blokhin et al19 demonstrated the benefit of melphalan in MM. Maas looked at corticosteroids in MM and determined that prednisone as a single agent produced significant decreases in serum globulin and an increase in haematocrit. Thereafter, Alexanian et al established the classic regimen of melphalan plus prednisone (MP) in a randomised trial of 183 patients, in which survival was six months longer with MP compared with melphalan alone.20 Melphalan and prednisolone remained the mainstay of myeloma treatment for decades.
Novel agents
The most significant advances in the treatment of MM have mostly occurred in the decade from 1998 to 2008. Thalidomide,21 bortezomib,22-24 and lenalidomide25 have emerged as highly active agents in the treatment of MM, each with a unique and interesting historical perspective. Even though thalidomide has been known for longer than the other two drugs, it is still considered a novel agent.
Thalidomide
Thalidomide was first introduced by Grünenthal, a German pharmaceutical company, as a sedative in 1957.26 By 1960 it was sold in more than 40 countries. It acquired a reputation as a popular drug for morning sickness in pregnancy. In 1961, Widukind Lenz, a German physician determined that thalidomide was associated with severe teratogenic malformations.27 By the end of 1961 thalidomide was taken off the market in most countries, however, almost 10,000 infants had already been affected.
Not long after its teratogenic properties were discovered, thalidomide was considered a possible treatment for cancer. In 1994, the spouse of an affected myeloma patient convinced Barlogie and colleagues at the University of Arkansas to initiate a compassionate-use trial of ‘antiangiogenic therapy’. As a consequence, thalidomide was tested. In a well-known, single-arm study by Barlogie et al,21 32% of patients responded to thalidomide, making it the first new drug with single-agent activity for myeloma in more than three decades. Several combination regimens that contain thalidomide were developed.
Bortezomib
Bortezomib is a boronic acid dipeptide, which inhibits proteasome activity and leads to cellular apoptosis. Malignant, transformed and proliferating cells are more susceptible to this effect. Preclinical studies demonstrated that bortezomib had potent cytotoxic and growth inhibitory effects.28 Bortezomib demonstrated striking anti-myeloma activity. Remarkable results from a phase II trial led to the approval of bortezomib by the US Food and Drug Administration (FDA) in May 2003.22
Lenalidomide
Lenalidomide belongs to a class of thalidomide analogues termed immunomodulatory drugs. The FDA approved lenalidomide plus dexamethasone in June 2006 for the treatment of MM in patients who have failed one prior therapy.29,30 Lenalidomide is also used in various combination regimens in order to obtain lasting remissions.
Transplant eligible versus transplant ineligible
Myeloma is considered to be a remitting/relapsing disorder where, typically, a patient will experience several relapses and require multiple lines of therapy. Whereas the combination of two drugs was the standard therapy in the 1960s through to the 1990s, the consensus now is a three- or four-drug combination therapy for initial treatment. However, much work is still needed to determine the best combination and the best sequence.
Patients who are not transplant candidates are frequently treated with standard alkylating agent therapy in combination with novel agents, eg. melphalan, prednisolone, bortezomib (MPV) or melphalan, prednisolone and thalidomide (MPT). The addition of these novel agents to the long-standing standard regimen of melphalan and prednisolone has produced remarkable increases in disease-free and overall survival.31,32
Another common approach is cyclophosphamide, bortezomib and dexamethazone, which, as a stem-cell sparing regimen, is also widely used as initial therapy in patients planned for autologous stem cell transplant.
Autologous stem cell transplant (ASCT)
In the early years of the development of stem cell transplantation, Thomas et al33 treated six patients, one of whom had MM, with total body irradiation or chemotherapy followed by an intravenous infusion of bone marrow cells. Barlogie demonstrated feasibility for high-dose therapy and autologous stem cell rescue in myeloma.34 Later, the role of ASCT in MM was established by the French IFM 90 trial35 and the British Medical Research Council (MRC) Myeloma VII trial.36
Today, every new patient is assessed as to their eligibility for transplantation. This is based on age, performance status, comorbidities and patient preferences. Although not curative, ASCT improves complete response rates and prolongs median overall survival in myeloma.17,35 The procedure-related mortality rate is low, 1-2%. The need for early ASCT in an era of new drugs is the most important clinical question in myeloma today. Ongoing studies will provide the answer, until then ASCT is deemed an essential requirement for eligible patients.
Allogeneic transplantation has had a limited role in selected patients and is not routinely considered. This fact has not significantly changed across the years.37
Maintenance
The advent of maintenance therapy has impacted the time to progression data for most MM patients able to tolerate ongoing therapy. Current evidence has shown lenalidomide to be a relatively safe and effective agent to reduce the risk of relapse in the post-transplant setting.38 More studies are awaited to determine the duration to which it can be used safely. However, an increased number of secondary cancers were observed in two trials of lenalidomide maintenance after autologous transplantation for myeloma,38,39 and in the non-transplant MM-015 trial by Palumbo et al.40 Palumbo argued that this slight risk (about 5%) was far outweighed by lenalidomide’s benefit in delaying progression of myeloma (about 75%).
Bortezomib has also been considered in ongoing studies for use as maintenance.41 Limitation is anticipated because of its associated peripheral neuropathy.
The future in myeloma treatment
As is the case in many oncological diseases, there is a large repertoire of drugs currently being tested. Some are very much in the experimental stage while others are in phase III trials around the world. An example of the latter is pomalidomide, which is the newest of the immunomodulatory drugs showing significant promise.42 Another new drug, elotuzumab, is a humanised monoclonal antibody to a protein called CS1 found to be highly expressed on plasma cells.43
Supportive measures
In addition to MM disease control, symptom management and supportive care measures have also taken precedence in recent decades leading to improvement in quality of life and, in turn, improved patient survival. Of particular interest is the role of bisphosphonates in reducing bone disease-related complications, as well as a demonstrated improvement in overall survival.44 Other notable developments include erythropoietin and immunoglobulin infusions.
Conclusion
Myeloma is a disease with an interesting history, not only from the point of view of treatment evolution but also with respect to developments in the management of side-effects and complications. Novel therapies have become the mainstay for MM with enviable remission rates. As our knowledge of disease pathophysiology expands so does the search for newer treatment modalities with fewer side-effects. The next few years promise to be a continuum of progress and promising advances that will no doubt go down in the history books of medicine.
References
Kyle RA, Rajkumar SV. Multiple myeloma. NEJM 2004; 351: 1860-1873
Bence Jones H. Chemical pathology. Lancet 1847; 2: 88-92
Bradwell AR, Carr-Smith HD, Mead GP et al. Highly sensitive, automated immunoassay for immunoglobulin free light chains in serum and urine. Clinical Chemistry. Am Ass Clin Chem 2001; 47(4): 673-680
Perlzweig WA, Delrue G, Geschicter C. Hyperproteinemia associated with multiple myelomas: report of an unusual case. JAMA 1928; 90: 755-757
Tiselius A. A new apparatus for electrophoretic analysis of colloidal mixtures. Trans Faraday Soc 1937; 33: 524
Grabar P, Williams CA. Methode permettant l’etude conjuguee des proprietes electrophoretiques et immunochimiques d’un melange de proteines; application au serum sanguine. Biochim Biophys Acta 1953; 10: 193-194
Wilson AT. Direct immunoelectrophoresis. J Immunol 1964; 92: 431-434
Waldenström J. Studies on conditions associated with disturbed gamma globulin formation (gammopathies). Harvey Lect 1961; 56: 211-231
Kyle RA. Personal communication to Gerard Crotty, 2012
Kyle RA. Monoclonal gammopathy of undetermined significance: natural history in 241 cases. Am J Med 1978; 64: 814-826
Durie BG, Salmon SE. A clinical staging system for multiple myeloma: Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer 1975; 36: 842-854
Greipp PR, San Miguel JF, Durie BG et al. International staging system for multiple myeloma. J Clin Oncol 2005; 23: 3412-3420
Dispenzieri A, Rajkumar SV, Gertz MA, et al. Treatment of newly diagnosed multiple myeloma based on Mayo stratification of myeloma and risk-adapted therapy (mSMART): Consensus Statement. Mayo Clin Proc 2007; 82: 323-341
Alwall N. Urethane and stilbamidine in multiple myeloma: report on two cases. Lancet 1947; 2: 388-389
Blokhin N, Larionov L, Perevodchikova N et al. Clinical experiences with sarcolysin in neoplastic diseases. Ann NY Acad Sci 1958; 68: 1128-1132
Alexanian R, Haut A, Khan AU et al. Treatment for multiple myeloma: combination chemotherapy with different melphalan dose regimens. JAMA 1969; 208: 1680-1685
Singhal S, Mehta J, Desikan R et al. Antitumor activity of thalidomide in refractory multiple myeloma. NEJM 1999; 341: 1565-1571
Richardson PG, Barlogie B, Berenson J et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. NEJM 2003; 348: 2609-2617
Richardson PG, Sonneveld P, Schuster MW et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. NEJM 2005; 352: 2487-2498
Rajkumar SV, Hayman SR, Lacy MQ, et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005; 106: 4050-4053
Richardson PG, Blood E, Mitsiades CS et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood 2006; 108: 3458-3464
Rajkumar SV. Thalidomide: tragic past and promising future. Mayo Clin Proc 2004; 79: 899-903
Lenz W. Thalidomide and congenital abnormalities. Lancet 1962; 1: 45-46
Hideshima T, Richardson P, Chauhan D et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001; 61: 3071-3076
Dimopoulos M, Spencer A, Attal M et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. NEJM 2007; 357: 2123-2132
Weber DM, Chen C, Niesvizky R et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. NEJM 2007; 357: 2133-2142
Facon T, Mary JY, Hulin C et al. Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomized trial. Lancet 2007; 370: 1209-1218
San Miguel JF, Schlag R, Khuageva NK et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. NEJM 2008; 359(9): 906-917
Thomas ED, Lochte HL, Jr, Lu WC et al. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. NEJM 1957; 257: 491-496
Barlogie B, Hall R, Zander A et al. High-dose melphalan with autologous bone marrow transplantation for multiple myeloma. Blood 1986; 67(5): 1298-1301
Attal M, Harousseau JL, Stoppa AM et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma: Intergroupe Francais du Myelome. NEJM 1996; 335: 91-97
Child JA, Morgan GJ, Davies FE et al. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. NEJM 2003; 348: 1875-1883
Lockhorst H et al, Inernational Myeloma Working Group Consensus Statement regarding the current status of stem-cell transplantaion for multiple myeloma. J Clin Oncol 2010; 28: 4521-30
Attal M, Lauwers-Cances V, Marit G et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. NEJM 2012; 366: 1782-1791
McCarthy PL, Owzar K, Hofmeister CC et al. Lenalidomide after stem-cell transplantation for multiple myeloma. NEJM 2012; 366(19): 1770-1781
Palumbo A, Hajek R, Delforge M et al. Continuous lenalidomide treatment for newly diagnosed multiple myeloma. NEJM 2012; 366(19): 1759-1769
Sonneveld P, Schmidt-Wolf IGH, van der Holt B et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: Results of the randomized phase III HOVON-65/GMMG-HD4 trial. J Clin Oncol 2012; 30(24): 2946-2955
Mikhael J, Rajkumar V, Roy V et al. Efficacy of pomalidomide plus low-dose dexamethasone in multiple myeloma patients despite previous use of lenalidomide. J Clin Oncol (ASCO Annual Meeting Abstracts) 2011; 29: (suppl; abst 8067)
Lonial S, Jakubowiak AJ, Jagannath S et al. A phase 2 study of elotuzumab in combination with lenalidomide and low-dose dexamethasone in patients with relapsed/refractory multiple myeloma. ASH Annual Meeting. Blood 2011; 118(21): abst 303
Morgan, GJ, Davies FE, Gregory WM et al. First-line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC Myeloma IX): a randomised controlled trial. Lancet 2010; 376(9757): 1989-1999
 (click to enlarge)
(click to enlarge)