The FORESIGHT Clinic: overseeing risk evaluation and surveillance of intestinal and gastric hereditary tumours
Surveillance of intestinal and gastric hereditary tumours
Dr Éanna Ryan, Research Registrars, St Vincent's University Hospital, Dublin, Dr Ben Creavin, Research Registrar, St Vincent's University Hospital, Dublin and Prof Des Winter, Consultant General and Colorectal Surgeon, St Vincent's University Hospital, Dublin
A cancer syndrome is a genetic condition in which an inherited mutation predisposes affected individuals to cancer development, often at a young age.1 Approximately 5-10% of all colorectal cancer (CRC) arises in this setting; while up to a third exhibit a moderate, familial pattern of inheritance, without an identified mutation.2 These patients benefit from increased surveillance, different adjuvant treatments, and preventative surgery.3-5 It has been recommended that individuals with known or suspected inherited cancer should be co-ordinated in specialised clinics.6 The appropriate management of such individuals through the use of registries and dedicated clinics results in a reduction in cancer incidence and mortality.7
The FORESIGHT Clinic
Genetic testing has grown from a niche specialty for rare disorders to one that has more routine clinical applications.8 Advances in technology have resulted in widely available, inexpensive next-generation sequencing (NGS) genetic screening panels for cancer syndromes. As a result, access to genetic testing is no longer the limiting factor in the management of these individuals. Ascertainment of family history, determination of cancer risk, education and counselling, provision for molecular testing (where appropriate), and specified follow-up for individuals based on their risk in a specialised environment is critical for these individuals. It is for these reasons that the FORESIGHT Clinic has been established at St Vincent’s University Hospital in Elm Park, Dublin 4.
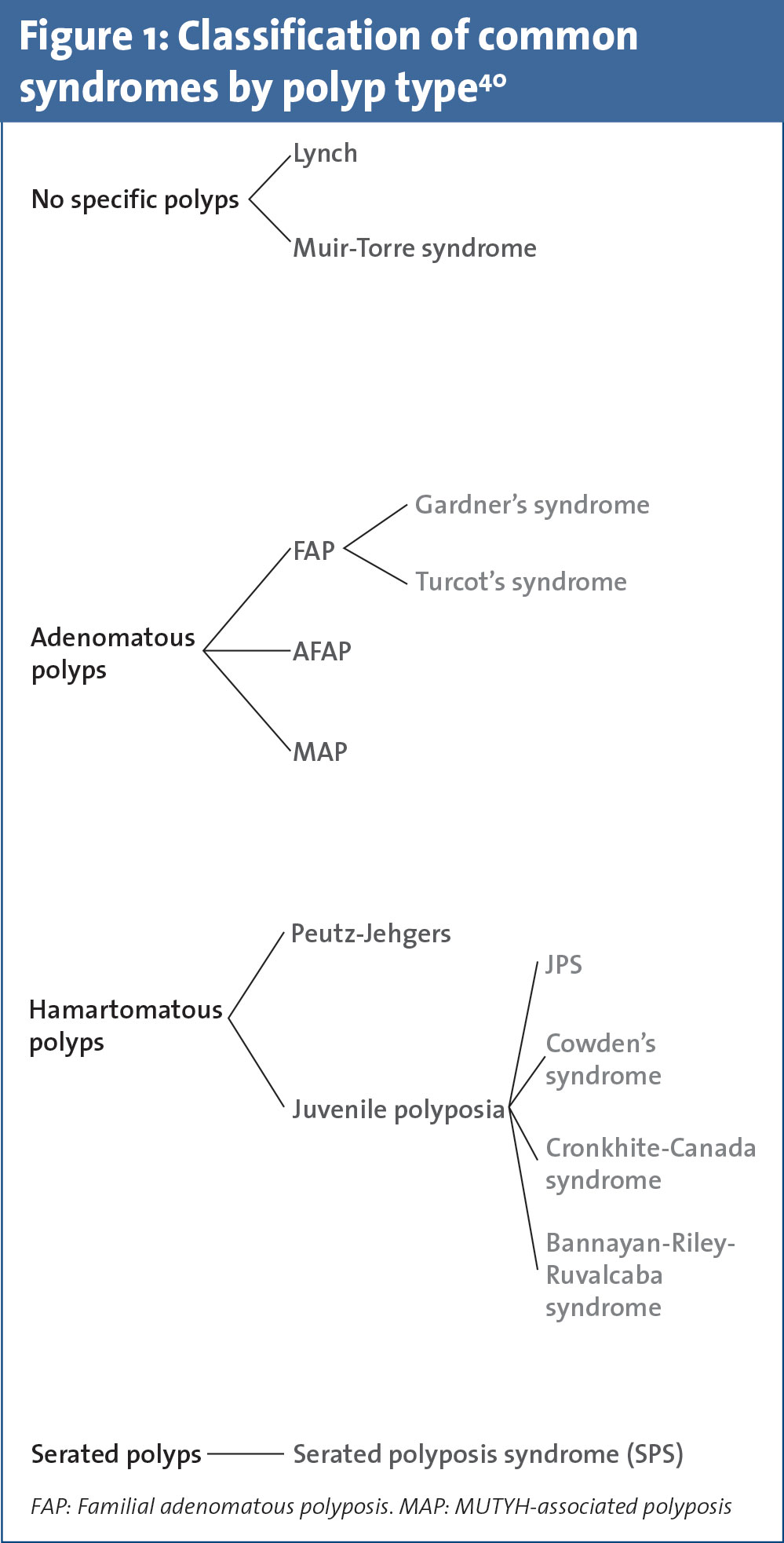
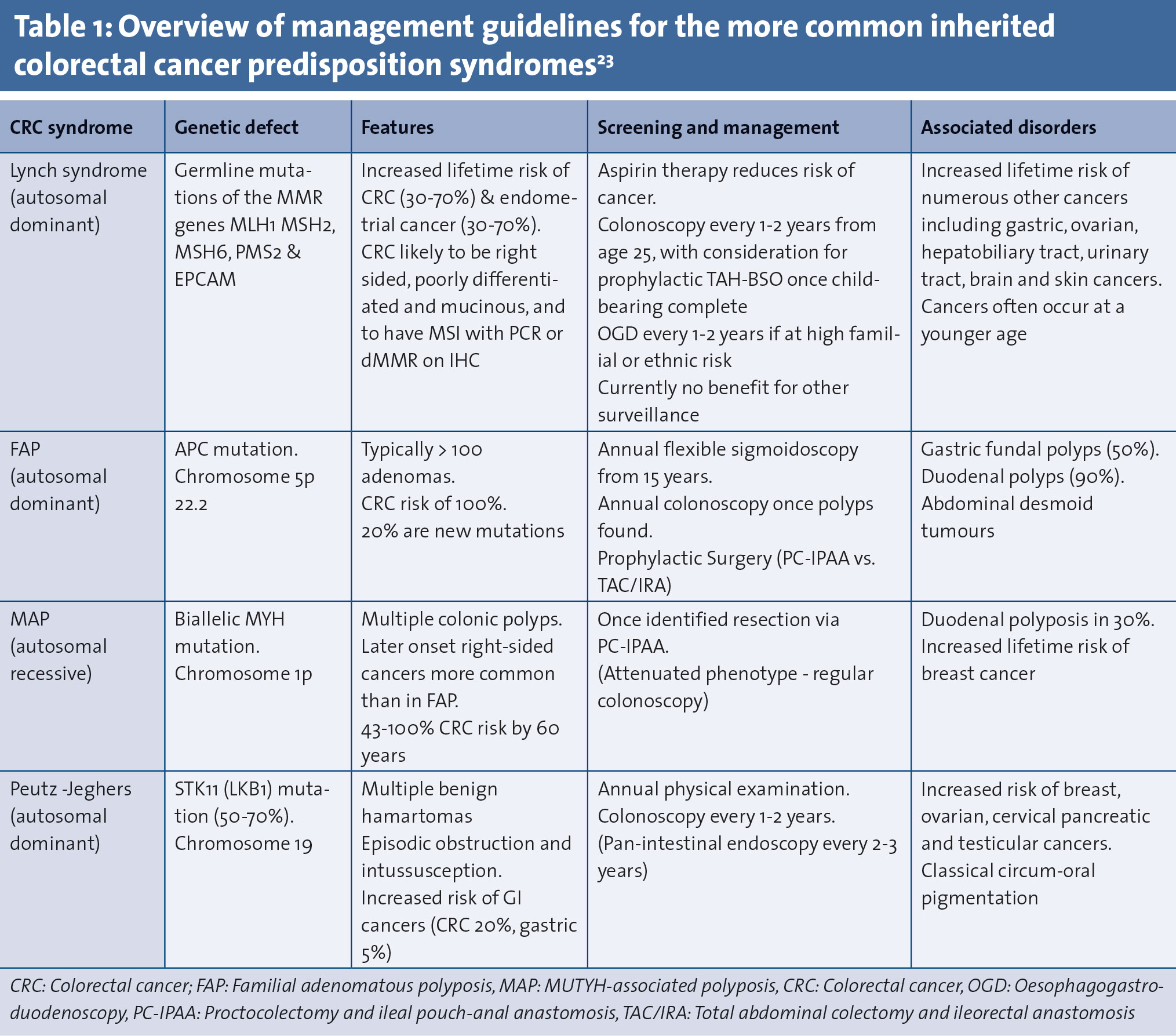
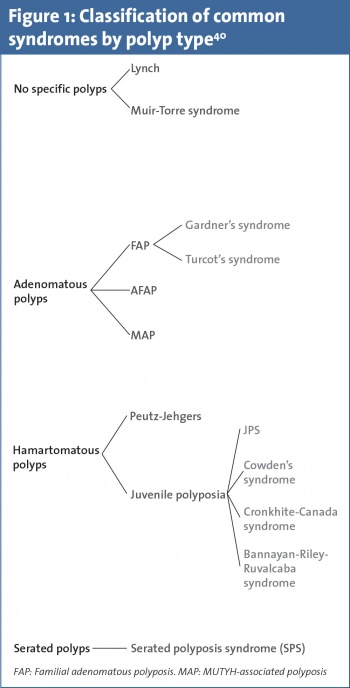
This specialised clinic involves a focused patient management programme incorporating a cancer syndrome registry, specialist nurses, dedicated gastroenterology and surgical consultants, as well as access to counselling, genetic testing and psychological support. In this review we have divided the most common CRC predisposition syndromes into two main groups, Lynch syndrome (formerly hereditary non-polyposis coli [HNPCC]) and the polyposis syndromes (see Figure 1 and Table 1). These individuals may present via three separate channels:
• An individual with a family or personal history warranting risk assessment
• An individual with a CRC that has tumoural- or patient-related characteristics warranting further investigation
• An individual with a known syndrome requiring life-long surveillance, risk reduction therapy and perhaps prophylactic surgery.
(click to enlarge)
(click to enlarge)
Assessment of high-risk patients
It is important to be aware that an individual with a significant family history of cancer may have a varying degree of risk depending on the underlying diagnosis. In some instances genetic testing may be necessary, with subsequent close endoscopic surveillance and/or prophylactic surgery. However, in other cases the patient may be found to carry no additional risk and may require no other follow up save that recommended for the general population. Therefore, the principal role of a specialised clinic should be to determine risk and to outline specified follow-up for that individual. The principle tool of risk assessment should be a comprehensive family history. The American Society of Clinical Oncology has recommended a minimum family history set (see Table 2).9 However, in busy clinical practice detailed histories are often difficult to obtain.10,11 This has led to the promotion of other methods to guide patient management. The Amsterdam II and Revised Bethesda criteria have been used to assess risk of Lynch syndrome (see Table 3).12-14 More recently, computer-based calculators have also become available.15-17
(click to enlarge)
Genetic testing
Where a CRC syndrome is suspected genetic and/or tumour analysis should begin with an affected patient or “proband”.18 Testing of relatives can be performed afterwards if a mutation is discovered. It is important that patients are made aware of their rights and that the implications of all possible results should be explained prior to testing.19 Testing may only take place with a person’s consent, in accordance with the Data Protection Acts. The perceived harmful effects of a positive test result, such as social stigmatisation and disadvantages relating to employment or health insurance, should be carefully discussed.20 Under Part 4 of the 2005 Disability Act, the results of a genetic test can’t be used in relation to insurance, a mortgage, a personal pension or employment in Ireland. However, there have been anecdotal reports that disclosure of genetic testing results is frequently made on proposal forms to insurance companies. Individuals should be explicitly instructed not to disclose this information. In the event of a genetic test result coming into the possession of an insurer, the genetic test result cannot be taken account by the insurer in any form.
The major difficulty in genetic testing may arise with result interpretation. A positive finding is helpful and patients can be counselled according to their risk. Relatives without a previously identified specific mutation (ie. a true negative) can be considered to have no increased risk and reassured accordingly. Problems may arise in patients with a suspected syndrome but in which no known mutation is found. If enough clinical suspicion exists to order genetic screening, a negative result must be interpreted with caution due to limitations of current technology and knowledge of the genes involved in heritable CRC.19 The use of NGS may also lead to a high rate of findings of variants of unknown significance (VUS).21,22 This is a mutation in which the extent of its role in cancer development is unknown. Therefore, a VUS has no established management recommendations. The absence of a living affected relative or access to suitable tumour tissue may also be problematic, as are circumstances where, following counselling, a relative declines testing.
Surveillance of high-risk patients
For patients with a suspected polyposis syndrome but no identified mutation, colonoscopic surveillance should commence as for patients with a known mutation. Findings at colonoscopy may be used to guide the rate of surveillance and if no polyps are detected over time, it may be prudent to increase the interval between scopes, as the patient gets older.23 Definitive guidance as to the optimal surveillance for patients without a recognised mutation or for those with VUS is lacking. These patients are often falsely reassured by a negative result and remain at risk despite failure to detect a specific mutation.24
Lynch syndrome
Lynch syndrome accounts for approximately 1-3% of all CRC.25 It is due to autosomal dominant mutations in the mismatch repair (MMR) genes MLH1, MSH2, MSH6 and PMS2.12,26-32 More recently, mutations in the epithelial cell adhesion molecule (EpCAM) gene have been implicated.33,34 Defective MMR genes results in CRC with microsatellite instability (MSI) and is characteristic of Lynch syndrome.35 Lynch syndrome patients carry an increased predisposition to CRC, endometrial, gastric, ovarian, hepatobiliary tract, urinary tract, brain and skin cancers, and these cancers often occur at a younger age.25,36-38 Lifetime incidence of CRC is reported between 35-80%.39,40 There is also an increased incidence of metachronous and synchronous colon cancers.31
Relying solely on age and family history based criteria inaccurately identifies eligibility for Lynch syndrome testing in 25%-70% of cases.12-14 Many institutions now advocate universal tumour screening via either polymerase chain reaction (PCR) for MSI or immunohistochemistry (IHC) for deficient MMR genes.41,42 The majority of MSI, however, occurs sporadically and is due to MLH1 promoter hypermethylation.43,44 The BRAF V600E mutation is present in approximately 60-70% of these sporadic cases.42 Therefore, if MLH1 loss is found; the tumour may be tested for either BRAF mutation or direct testing of MLH1 promoter hypermethylation to help distinguish between Lynch syndrome and sporadic CRC.25
A patient with hypermethylated MLH1 and/or a BRAF mutation is very unlikely to be Lynch syndrome.45 Patients with absent MLH1 and wild type BRAF, and those with absent MSH2, MSH6 or PMS2 should be offered testing.
Surveillance and management of lynch syndrome mutation carriers
There is no role for prophylactic colectomy in Lynch syndrome.4,46 Prophylactic hysterectomy and bilateral salpingo-oophorectomy should be considered in women who have completed childbearing or after age 40 years as it significantly reduces the risk of both endometrial and ovarian cancer.4,46 Recommended surveillance includes annual colonoscopy starting at age 25 (or five years prior to the youngest affected family member if < 30 years) and two-yearly upper endoscopy starting at 30 in families with gastric cancer or at high ethnic risk.
While some guidelines recommend annual per vaginal examination, pelvic ultrasound, Ca-125 analysis, and urinalysis, there is currently no evidence for benefit.4,47 The CAPP2 trial showed a significant reduction in CRC and other cancers among Lynch syndrome patients who received aspirin.48,49
Management of patients with cancer due to Lynch syndrome
The European Hereditary Tumour Group recommends total colectomy due to risk of recurrence following segmental colectomy.4 A recent meta-analysis found metachronous CRC occurred more frequently after segmental colectomy (23.5% versus 6.8%) but there was no survival difference.5 There is also evidence that total colectomy is associated with equivalent quality of life despite poorer functional outcomes.50 Nevertheless, segmental colectomy is often performed if the diagnosis is not recognised prior to operation, and may be appropriate in patients with known Lynch syndrome who have significant comorbidities, metastatic disease or poor anal muscle tone.51
Patients who present with an index rectal cancer should be considered for proctocolectomy. Recommended surveillance includes annual endoscopy of residual colon and/or rectum.51-53
Intestinal polyposis syndromes
Familial adenomatous polyposis
Familial adenomatous polyposis (FAP) is an autosomal dominant disorder occurring due to APC mutations.54 Patients frequently present in their thirties with adenomatous polyp burdens of >100, predominantly in the left colon.55 Attenuated Familial Adenomatous Polyposis (AFAP) is similarly associated with APC. Patients present somewhat later and <100 polyps are usually seen with a more proximal distribution.56 MYH-Associated Polyposis (MAP) is an autosomal recessive condition due to biallelic MUTYH mutations.57 It is most commonly characterised by 20-99 adenomatous polyps.58
Syndromes associated with multiple hamartomas
Peutz-Jeghers’ syndrome is characterised by multiple gastrointestinal (GI) hamartomas and muco-cutaneous hyper-pigmentation. Mutations in STK11 are seen in 50-70% of cases. Patients have increased risk of developing several GI cancers over their lifetime.59 Germline mutations in SMAD 4 and BMPR1A occur in approximately 50% of patients with juvenile polyposis syndrome (JPS).60 The finding of GI hamartomas in patients with a strong family history of GI cancers should prompt this disorder to be suspected. Cowden syndrome is related to germline mutations in the PTEN gene. Colonic hamartomas are seen, however, adenomas and hyperplastic polyps may also be identified.61 Cowden syndrome is more frequently associated with other cancer types such as breast, endometrial, thyroid and renal cancers.62
Serrated polyp syndrome
Serrated (formerly known as hyperplastic) polyp syndrome (SPS) is a rare condition with no known specific germline mutation.63 Currently there is no known specific germline mutation associated with SPS. However, if adenomas are also present, testing for MUYTH mutation should be considered as overlap with MAP has been described.64
Surveillance of patients with polyposis syndromes
Due to the extreme polyp burden, the primary role of endoscopy in classical FAP is limited to monitoring polyp progression.65 Endoscopy should begin at puberty or earlier if symptoms occur. Initially a yearly flexible sigmoidoscopy is adequate. Yearly colonoscopy becomes mandatory once polyps develop. Polyps associated with AFAP usually occur later in life than classical FAP.66 Full colonoscopy is the required due to the more proximal location of the polyps. Surveillance starts at puberty and must continue every 1-2 years depending on polyp burden however, due to the comparative late onset of polyps in contrast to FAP, surveillance can justifiably be delayed until later in the teenage years.67 Surveillance in the less common polyposis syndromes (MAP, hamartomatous syndromes, SPS) is mainly centred on polyp burden at initial colonoscopy.68 In the case of JPS, known mutation carriers should undergo an initial colonoscopy at puberty. Surveillance in others should commence in the mid-late teens. Colonoscopy should be repeated every 1-2 years until polyps develop. If polyps can be controlled endoscopically, yearly colonoscopy is appropriate.
Surgical management of polyposis syndromes
Development of CRC in FAP is inevitable without prophylactic surgery. Urgent surgery should be considered in those with intolerable symptoms of polyposis, rapidly expanding polyp numbers, increasing number of polyps > 1cm, evidence of high-grade dysplasia, and poor compliance with yearly surveillance.69 The best procedure for risk reduction is proctocolectomy with ileal pouch-anal anastomosis. It may be reasonable in patients with low polyp burden in the rectum (<20) to undergo total abdominal colectomy with ileorectal anastomosis (TAC/IRA) as it has better functional outcomes.69 However, there is data that demonstrates that even with ideal surveillance, advanced rectal cancers can develop in FAP patients who undergo rectal-preserving surgery.70 Ultimately the decision should be made on a case-by-case basis. If the rectum is left in situ, six month-yearly proctoscopy is required to provide surveillance. Pouch surveillance can be performed every 1-3 years depending on polyp growth.
In contrast, AFAP does not unavoidably lead to cancer but risk does increase with age. The mean age of cancer diagnosis with AFAP has been reported as 58 years, with a 69% risk of CRC development by 80 years.56 The more proximal poly burden with relative rectal sparing makes TAC/IRA a good option. However, patients with a low polyp burden may be managed endoscopically. In the less common polyposis syndromes consideration for surgery generally follows the same guidelines as for AFA
Conclusion
Recent technological advancements have resulted in easily available and affordable genetic screens. Colorectal specialists and gastroenterologists, rather than medical geneticists, now have the opportunity to become the main providers of care for patients with or suspected to have CRC syndromes. It is important that other medical practitioners are aware as to the heritable nature of CRC. While there has been a strong recommendation for the management of these patients to be co-ordinated through specialised clinics and cancer registries this has yet to occur in Ireland.6 Dedicated clinics are required to develop management guidelines and to improve patient outcomes. It is for these reasons, that the FORESIGHT Clinic has been established at St Vincent’s University Hospital.
References
Garber, JE and K Offit, Hereditary cancer predisposition syndromes. J Clin Oncol 2005; 23(2): 276-92
Burt R. Inheritance of Colorectal Cancer. Drug discovery today. Disease mechanisms 2007; 4(4): 293-300
Le DT, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015; 372(26): 2509-20
Vasen HF, et al. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium “Care for CMMR-D” (C4CMMR-D). J Med Genet 2014; 51(5): 283-93
Heneghan HM, Martin ST, Winter DC. Segmental vs extended colectomy in the management of hereditary nonpolyposis colorectal cancer: a systematic review and meta-analysis. Colorectal Dis 2015; 17(5): 382-9
Collins V, et al. Assessment of education and counselling offered by a familial colorectal cancer clinic. Clin Genet 2000; 57(1): 48-55
Barrow, P., et al., Systematic review of the impact of registration and screening on colorectal cancer incidence and mortality in familial adenomatous polyposis and Lynch syndrome. Br J Surg 2013; 100(13): 1719-31
Katsanis SH, Katsanis N. Molecular genetic testing and the future of clinical genomics. Nat Rev Genet 2013; 14(6): 415-26
Lu KH, et al. American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers. J Clin Oncol 2014; 32(8): 833-40
Wilson BJ, et al. Systematic review: family history in risk assessment for common diseases. Ann Intern Med 2009; 151(12): 878-85
Mitchell RJ, et al. Accuracy of reporting of family history of colorectal cancer. Gut 2004; 53(2): 291-5
Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007; 50(1): 113-30
Peterlongo P, et al. MSH6 germline mutations are rare in colorectal cancer families. Int J Cancer 2003; 107(4): 571-9
Gologan A, Sepulveda AR. Microsatellite instability and DNA mismatch repair deficiency testing in hereditary and sporadic gastrointestinal cancers. Clin Lab Med 2005; 25(1): 179-96
Chen S, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. Jama 2006; 296(12): 1479-87
Kastrinos F, et al. The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology 2011; 140(1): 73-81
Green RC, et al. Prediction of Lynch syndrome in consecutive patients with colorectal cancer. J Natl Cancer Inst 2009; 101(5): 331-40
Stoffel EM. Screening in GI Cancers: The Role of Genetics. J Clin Oncol 2015; 33(16): 1721-8
Riley BD, et al. Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns 2012; 21(2): 151-61
Hodgson S, et al. Cancer genetics services in Europe. Dis Markers 1999; 15(1-3): 3-13
Yurgelun MB, et al. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015; 149(3): 604-13.e20
Selkirk CG, et al. Cancer genetic testing panels for inherited cancer susceptibility: the clinical experience of a large adult genetics practice. Fam Cancer 2014; 13(4): 527-36
Genetic/Familial High-Risk Assessment: Colorectal National Comprehensive Cancer Network (NCCN) guidelines for detection, prevention and risk reduction, 2015. (Version 1.2015)
Grover S, et al. Colorectal cancer risk perception on the basis of genetic test results in individuals at risk for Lynch syndrome. J Clin Oncol 2009; 27(24): 3981-6
Vasen HF, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44(6): 353-62
Aarnio M, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999; 81(2): 214-8
Gazzoli I, et al. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res 2002; 62(14): 3925-8
Takemoto N, et al. The correlation of microsatellite instability and tumor-infiltrating lymphocytes in hereditary non-polyposis colorectal cancer (HNPCC) and sporadic colorectal cancers: the significance of different types of lymphocyte infiltration. Jpn J Clin Oncol 2004; 34(2): 90-8
Koinuma K, et al. Mutations of BRAF are associated with extensive hMLH1 promoter methylation in sporadic colorectal carcinomas. Int J Cancer 2004; 108(2): 237-42
Lagerstedt Robinson K, et al. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst 2007; 99(4): 291-9
Lynch HT, et al. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet 2009; 76(1): 1-18
Bonadona V, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. Jama 2011; 305(22): 2304-10
Ligtenberg MJ, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet 2009; 41(1): 112-7
Kovacs ME, et al. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 2009; 30(2): 197-203
Yamamoto H, Imai K. Microsatellite instability: an update. Arch Toxicol, 2015; 89(6): 899-921
Vasen HF. Clinical description of the Lynch syndrome [hereditary nonpolyposis colorectal cancer (HNPCC)]. Fam Cancer 2005; 4(3): 219-25
Vasen HF, Boland CR. Progress in genetic testing, classification, and identification of Lynch syndrome. Jama 2005; 293(16): 2028-30
Abdel-Rahman WM, Mecklin J, Peltomaki P. The genetics of HNPCC: application to diagnosis and screening. Crit Rev Oncol Hematol 2006; 58(3): 208-20
Sehgal R, et al. Lynch syndrome: an updated review. Genes (Basel) 2014; 5(3): 497-507
Sammour T, et al. Familial colorectal cancer syndromes: an overview of clinical management. Expert Rev Gastroenterol Hepatol 2015; 9(6): 757-64
Snowsill T, et al. A systematic review and economic evaluation of diagnostic strategies for Lynch syndrome. Health Technol Assess 2014; 18(58): 1-406
Palomaki GE, et al. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med 2009; 11(1): 42-65
Imai K, Yamamoto H. Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis 2008; 29(4): 673-80
Kim JH, Kang GH. Molecular and prognostic heterogeneity of microsatellite-unstable colorectal cancer. World J Gastroenterol 2014; 20(15): 4230-43
Domingo E, et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet 2004; 41(9): 664-8
Schmeler KM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med 2006; 354(3): 261-9
Auranen A, Joutsiniemi T. A systematic review of gynecological cancer surveillance in women belonging to hereditary nonpolyposis colorectal cancer (Lynch syndrome) families. Acta Obstet Gynecol Scand 2011; 90(5): 437-44
Burn J, et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N Engl J Med 2008; 359(24): 2567-78
Burn J, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011; 378(9809): 2081-7
Haanstra JF, et al. Quality of life after surgery for colon cancer in patients with Lynch syndrome: partial versus subtotal colectomy. Dis Colon Rectum 2012; 55(6): 653-9
Jasperson KW, et al. Hereditary and familial colon cancer. Gastroenterology 2010; 138(6): 2044-58
Balmana J, et al. Familial risk-colorectal cancer: ESMO Clinical Practice Guidelines. Ann Oncol 2013; 24 Suppl 6: vi73-80
Schlussel AT, et al. The evolution of colorectal cancer genetics-Part 1: from discovery to practice. J Gastrointest Oncol 2014; 5(5): 326-35
Vasen HF. When should endoscopic screening in familial adenomatous polyposis be started? Gastroenterology 2000; 118(4): 808-9
Burt RW, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology 2004; 127(2): 444-51
Al-Tassan N, et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet 2002; 30(2): 227-32
Sieber OM, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 2003; 348(9): 791-9
Beggs AD, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010; 59(7): 975-86
Chow E, Macrae F. A review of juvenile polyposis syndrome. J Gastroenterol Hepatol 2005; 20(11): 1634-40
Heald B, et al. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology 2010; 139(6): 1927-33
Mester J, Eng C. Cowden syndrome: recognizing and managing a not-so-rare hereditary cancer syndrome. J Surg Oncol 2015; 111(1): 125-30
Rex DK, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol 2012; 107(9): 1315-29; quiz 1314, 1330
Boparai KS, et al. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology 2008; 135(6): 2014-8
Gibbons DC, et al. Colorectal cancer: no longer the issue in familial adenomatous polyposis? Fam Cancer 2011; 10(1): 11-20
Friedl W, et al. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut 2001; 48(4): 515-21
Stoffel EM, Mangu PB, Limburg PJ. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology clinical practice guidelines. J Oncol Pract 2015; 11(3): e437-41
van Lier MG, et al. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol 2010; 105(6): 1258-64; author reply 1265
Kalady MF, Church JM. Prophylactic colectomy: Rationale, indications, and approach. J Surg Oncol 2015; 111(1): 112-7
Vasen HF, et al. Decision analysis in the surgical treatment of patients with familial adenomatous polyposis: a Dutch-Scandinavian collaborative study including 659 patients. Gut 2001; 49(2): 231-5
 (click to enlarge)
(click to enlarge)