With the advent of new approaches and therapies in cystic fibrosis management come new challenges for the multidisciplinary team
Dr Hisham Ibrahim, Specialist Registrar, Cork University Hospital, Cork, Dr David Morrissy, Research Registrar, Cork University Hospital, Cork and Prof Barry J Plant, Consultant Respiratory Physician and Director of the Cork Adult CF Centre, Cork University Hospital, Cork
Cystic fibrosis (CF) is a progressive, systemic, life threatening, autosomal recessive disease that is caused by mutations in the CFTR gene. It is the most common autosomal recessive life reducing condition in Ireland with approximately one in 1,600 births affected. Approximately one in 19 individuals carry the defective gene in Ireland. A study in 2008 suggested that the mean prevalence in 28 EU countries was 0.737/10,000, while the Republic of Ireland had a documented prevalence of 2.98.1
Based on European registry data it has been predicted that by 2025 the number of patients with CF in western European countries will increase by approximately 50% (20% increase in the paediatric population and 75% in the adult population).2
Classically CF presents with recurrent meconium ileus in the newborn, failure to thrive and recurrent respiratory tract infections in infants or older children. It is commonly treated by respiratory physicians and nursing staff with specialist training in respiratory medicine. That said cystic fibrosis is a systemic disease as the cystic fibrosis transmembrane conductance regulator (CFTR) protein is expressed ubiquitously in the body. Therefore, additional supports are needed from endocrinology, gastroenterology, genitourinary, reproductive medicine and mental health to mention but a few (see section below on CF management).
In order to understand cystic fibrosis as a disease, one must have an appreciation of its basic pathophysiology. It is a condition caused by defective and/or the deficiency of CFTR protein (an anion channel present on epithelial membranes) at cell surfaces. This protein is primarily responsible for the transport of chloride and bicarbonate across epithelial cell membranes. In the lung, disturbed flow of chloride and sodium results in hyper-concentrated viscoid secretions, associated mucus build-up, infection, inflammation and progressively worsening lung function. Similarly, the extrapulmonary features of the disease can be attributed to the deficiency or defective nature of the CFTR protein located at the cell surfaces of other organ systems.3
The CFTR protein is coded for by the CFTR gene located on the long arm of chromosome 7 at position q31.2. It was identified in 1989 and at present, approximately 2,000 gene mutations have been identified, the most common being represented by the phe508del mutation. An individual must have a disease-causing mutation present at each allele on both chromosomes to be affected. The mutations can be classified broadly into one of six subclasses (see Figure 1) and recently genotype has had increasing clinical importance as certain personalised therapies have clinical importance on specific subclasses.
Figure 1: Overview for CFTR mutations and potential therapeutic approaches(click to enlarge)
A patient’s genotype dictates whether a patient is deficient in the CFTR protein or the CFTR protein that is expressed at their cell surfaces is defective, or both. This is the focus of much research recently and has resulted in the development of a new and expanding group of agents, known collectively as CFTR modulators, which are offering new therapeutic options directed at the primary mutations.
Survival is improving in CF. Traditionally a disease of childhood, now in Ireland over 50% of the population is adult and UK data supports that if born in the year 2000 the predicted median survival is greater than 50 years.4
CF diagnosis
Diagnosis of CF is made based on both of the following criteria:5
Symptoms consistent with CF in at least one organ system, or positive new-born screen or genetic testing for siblings of patients with CF
Evidence of CFTR dysfunction such as elevated sweat chloride, presence of two disease causing mutations on genetic analysis or abnormal nasal potential differences on testing.
A patient diagnosed with CF may report a variety of symptoms, however cough, shortness of breath and increased sputum production are routinely reported and assessed throughout the life span of a CF patient.
Symptoms vary depending on the age/severity profile of the patient at diagnosis. Newborns present with meconium ileus. Infants typically present with one or more of the following symptoms: recurrent respiratory symptoms and/or failure to thrive. Adult diagnosis is less common and is often atypical in its presentation, including recurrent pancreatitis, pancreatic insufficiency, infertility or bronchiectasis. In Ireland the newborn screening programme for CF commenced in 2012. Therefore, a diagnosis of CF must remain in the differential when assessing both symptomatic children and adults who were born prior to this date. Newborn screening consists of an initial measurement of serum immunoreactive trypsinogen (IRT), whose levels are elevated in a newborn with CF and a subsequent DNA analysis whereby a sample is tested for the presence of specific gene mutations.5
Sweat testing plays a crucial role in both the diagnosis and more recently in the response to treatment when commenced on CFTR modulator treatment. A sweat chloride result of ≤ 29mmol/l is regarded as normal. A sweat chloride ≥ 60mmol/l is regarded as abnormal and sufficient to diagnose CF in patients with clinical symptoms.5 Positive results should be further evaluated with CFTR mutation analysis and repeat sweat testing. A sweat chloride result > 29mmol/l and ≤59mmol/l suggests an intermediate result and calls for further investigation. DNA analysis can confirm a diagnosis by detecting the presence of two disease causing mutations on both alleles.
Approximately 2,000 gene mutations associated with CF have been identified since the identification of the CFTR gene in 1989. 45% of patients are homozygous for the most common gene mutation F508del worldwide, though this is higher in Ireland.6
CF management
CF management is under the remit of a multidisciplinary team (MDT) focusing on pulmonary exacerbation avoidance and screening of extra pulmonary manifestations in an increasing aged population.
While recently enormous strides have been made in the development of novel treatment options, including CFTR modulators, a number of more established principles continue to form foundation and the bulk of the work regarding the management of CF patients.
The CF MDT is made up of a lead clinician traditionally with a respiratory background, dedicated clinical nurse specialists, a dedicated physiotherapist, a dedicated dietitian and a dedicated psychologist, with the support of additional medical input from other services. The focus of the MDT depends on the age profile of the patient.
The goal of MDT treatment is to preserve lung function, minimise pulmonary exacerbations and screen for extrapulmonary manifestations such as diabetes. Preservation of lung function depends on good airway clearance techniques, complemented by the use of mucolytics5 (dornase alpha and hypertonic saline) to maximise pulmonary toilet, as well as maintaining nutritional status with dietitian support and for most pancreatic enzymes to ensure adequate nutritional absorption.
For the majority of patients, the natural history of CF involves the acquisition and potential colonisation of Pseudomonas aeruginosa in the lung which is associated with accelerated long term significant clinical decline. Delaying/eradication of P. aeruginosa is associated with improved survival. A series of eradication protocols exist which include the use of various inhaled antibiotics including colomycin, tobramycin and aztreonam, which are administered in a specific regimen.5 In those that remain positive, long-term suppression of P. aeruginosa with inhaled antibiotics decreases exacerbation frequency. Additionally, low dose macrolide therapy (azithromycin) is used in those who remain culture positive as it has been shown to reduce exacerbation frequency as well as improve lung function. With each exacerbation, CF patients have a 20-25% chance of not recovering their lung function to its pre-exacerbation level,7 making exacerbation avoidance critical to long-term wellbeing.
It is the opinion of the authors that the mantra of the CF MDT should be ‘exacerbation zero’.
New therapeutic focuses in CF: It’s all about CFTR modulators
New therapeutic focuses in CF is all about CFTR modulators. Cell surface CFTR protein function restoration has been tried using different therapeutic strategies, but the most successful therapeutic strategy to date are the CFTR potentiators and correctors.
Gene therapy
Since the discovery of the cystic fibrosis gene in 1989, several clinical trials investigated the concept of restoring the CFTR protein function by gene therapy, either via replacing the dysfunctional gene through adding the wild-type CFTR DNA into human cells along with a promoter to allow for long-term expression, or using gene editing strategy that removes and/or replaces the mutated sequence. But despite extensive studies in this field, there has been no positive outcome to date. A recent randomised controlled trial investigated gene therapy using a plasmid DNA encoding the CFTR gene complexed with cationic liposome in patients with cystic fibrosis; the outcome was associated with a stabilisation of lung function in the treatment arm compared with a decline in the placebo group.8 Difficulties of gene therapy have been attributed to:
Finding the suitable vector to transport the DNA into the host cell, along with promoters to allow for long term expression
The host immune response to the vector.
Efforts to overcome these hurdles are still ongoing, and the UK CF Gene Therapy Consortium (GTC) is currently investigating an alternative viral vector (Wave 2 products, using lentivirus to deliver the CF DNA).9
CFTR potentiators and correctors
The first successful CFTR potentiator was ivacaftor, which was initially used to target class III mutation with defective channel regulation. The result from phase III clinical trials investigating the efficacy of ivacaftor in adolescents/adults (STRIVE trial)10 and children (ENVISON)11 with G551D-CFTR mutation highlighted the potential benefit of this approach. In the STRIVE study at week 24 the forced expiratory volume in 1 second (FEV1) percentage predicted showed a statistically significant treatment effect of 10.6% points (p < 0.001), together with 55% reduction in the risk of pulmonary exacerbation (p = 0.001), and significant improvement in nutritional status, sweat chloride and patient reported outcomes. The ENVISION study had similar outcomes. Subsequent studies also highlighted the role of ivacaftor as a monotherapy in the setting of other gating, for example R117H mutation.12
In a clinical trial cohort, the long-term impact of ivacaftor has been proved in the roll over trial (PERSIST)13 where improvements in FEV1 reported during STRIVE were sustained for an additional 12 weeks beyond the initial 48-week study period. Our group highlighted the benefit of ivacaftor in a real-world cohort with significant improvement in the extent of mucus plugging, peri-bronchial thickening and total Bhalla bronchiectasis score using ultra low dose CT scan suggesting enhanced mucus clearance, and showed increased lung microbiota diversity.14
With more complex class II CFTR protein trafficking mutation, ie. phe508del mutation, dual therapy has been required to demonstrate a clinical effect. In the TRAFFIC and TRANSPORT trials,15 the CFTR corrector lumacaftor in combination with the CFTR potentiator ivacaftor were studied in patient homozygous for phe508del (CFTR protein trafficking defect), results were very encouraging in both studies, FEV1 improvements were observed as early as day 15 and were sustained through 24 weeks with an improvement ranged from 2.6 to 4.0 percentage points (p < 0.001 for all comparisons). Perhaps more importantly is that there was a 35% reduction in the exacerbation’s frequency at 24 weeks. Adverse events, including bronchospasm and drug-to-drug interactions, led to discontinuation of therapy in some patients. A new agent/CFTR corrector, namely tezacaftor, was introduced in combination with ivacaftor and clinical trials using tezacaftor-ivacaftor showed comparable results to lumacaftor-ivacaftor, however adverse events and drug-drug interactions were less common.16
Most recently, triple combination therapy for people with cystic fibrosis using next-generation correctors, VX-659 and VX-445 in addition to tezacaftor-ivacaftor have advanced into phase III development (ongoing studies), based on initial phase II data17,18 that showed mean absolute improvements in percent predicted forced expiratory volume in one second (ppFEV1) of up to 13.3 and 13.8 percentage points from baseline through four weeks of treatment for the triple combination regimens with VX-659 or VX-445, respectively, in people who have one F508del mutation and one minimal function mutation (F508del/Min).
These recent studies offer a potential benefit to more than 90% of the CF population if phase III studies are successful.
Read-through agent
Several clinical trials investigated ataluren read‑through agent in class 1 nonsense mutation. The Ataluren Confirmatory Trial (ACT CF) in nonsense mutation cystic fibrosis (nmCF) did not achieve its primary or secondary outcome.19
Challenges in CF
As we get better at treating lung disease in CF, extrapulmonary manifestations will require greater attention.
Multisystem involvement in CF
In 1938, cystic fibrosis was diagnosed as a separated disease from coeliac disease; it was considered as a pancreatic disease in infants and children who died with malnutrition. In 1950 with the advent of pancreatic enzyme supplementation, pulmonary disease became the dominant clinical problem.
As CFTR receptors are expressed ubiquitously, and with improved survival we are seeing a wide range of clinical problems including: CF-related diabetes (CFRD), renal disease (acute kidney injury and chronic kidney disease), metabolic bone disease, a higher prevalence of gastrointestinal cancer than age matched populations, drug allergies and toxicities, and other complications post lung transplant, including chronic kidney disease (CFRD) and increased cancer risk.20
Psychological challenges in CF
The clinical manifestations of CF start very early in life, and children born with CF and their families are faced with the challenges imposed by the chronic progressive nature of CF since birth and the burden of the disease. Patients with CF are often on multiple maintenance therapies, which is very demanding despite the improved outcomes. Prevalence of depression ranges from 8-29% in children with CF and from 13-33% in adults with CF. Additionally, rates of depression of 20-35% have been reported among caregivers of people with CF and this has been associated with increased prevalence of anxiety/depression in a study of children aged 7 to 11 years with CF.21,22
Screening of psychological symptoms, and much more focus on the psychological support and psychological health of patients with CF and their families, is required to maximise clinical outcome and patients’ quality of life.
Compliance to medications
Multiple studies have shown that poor adherence to medications is associated with increased hospitalisation, frequent exacerbations, lung function deterioration and worse overall health outcomes. Estimated adherence to medications in CF patients based on previous studies is approximately 50%, and recent studies in CFTR modulators showed self-reported adherence to medications is 100% compared to electronic monitoring of bottle opening rate of only 66%.23
Poor adherence to conventional therapy in CF and novel medications has a negative impact on disease progression, overall health outcome and quality of life. More focus is required on promoting adherence and simplifying delivery of existing medications, for example, a rapid delivery system for inhaled antibiotics.
Adherence to existing medications is as important as developing new medications in CF.
Drug interactions
In parallel with improving median survival, developing novel therapy for patients with CF and polypharmacy, drug-to-drug interaction is an emerging challenge. Furthermore, the multisystemic nature of disease that affects the absorption, distribution, metabolism, excretion and pharmacodynamics of drugs, sometimes unpredictably, will further complicate this.
Although, the impact of polypharmacy on drug-to‑drug interaction have been well studies among the general populations, there are very limited data on drug-to‑drug interaction among aging CF populations. Further studies are required to examine the potential impact of clinically significant drug-to‑drug interaction between novel therapy in CF and other medications.
Reproductive health
With improved survival many patients with CF are considering parenthood. In 2017 US Registry data documented 273 pregnancies in women with CF.24 While altered hydroelectrolytic contents of uterine secretions and abnormal viscosity of cervical mucus may induce barriers to conception, many women with CF are able to conceive spontaneously. Therefore, reproductive health advice is increasingly important including access to safe and efficient contraceptive advice unless planning a pregnancy. In this situation genetic counselling, potential complications and risks of pregnancy (particularly in severe disease) needs to be addressed and where applicable assisted fertility discussed in detail to allow for informed and best outcomes for mother and child.25
For men with CF, more than 95% are infertile due to an absence of the vas deferens. Semen analysis is an important service for adult males with CF and should be made available. Assisted reproduction techniques can be employed to facilitate pregnancy for them and their partners with an increasing rate of success in specialist centres.26
Conclusion
With the meticulous use of traditional therapies through a CF MDT, continued improved survival and quality of life for patients with CF is a reality in Ireland. The advent of new personalised approaches/therapies are improving outcomes further with a new era for CF care in Ireland a reality. The face of CF is changing with new pulmonary and extra-pulmonary challenges where engagement with the Irish medical community beyond the CF MDT will become even more important.
Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J 2007; 29(3):522-6
Smyth AR, Bell SC, Bojcin S et al. European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. J Cyst Fibros [Internet] 2014; 13(Suppl 1):23-42. http://dx.doi.org/10.1016/j.jcf.2014.03.010
The Clinical and Functional TRanslation of CFTR (CFTR2) [Internet]. Available from: http://cftr2.org
Sanders DB, Bittner RCL, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med 2010; 182(5):627-32
Alton EWFW, Armstrong DK, Ashby D et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med 2015; 3(9):684-91
Care S. The UK Cystic Fibrosis Gene Therapy Consortium, Boehringer Ingelheim, Imperial Innovations and Oxford BioMedica Announce New Partnership to Develop First-In-Class Gene Therapy for Cystic Fibrosis. 2018
Ramsey BW, Davies J, McElvaney NG et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N Engl J Med [Internet] 2011 Nov 3; 365(18):1663-72. http://www.nejm.org/doi/abs/10.1056/NEJMoa1105185
Davies JC, Wainwright CE, Canny GJ et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 2013; 187(11):1219-25
Moss RB, Flume PA, Elborn JS et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: A double-blind, randomised controlled trial. Lancet Respir Med [Internet]. 2015;3(7):524-33. http://dx.doi.org/10.1016/S2213-2600(15)00201-5
McKone EF, Borowitz D, Drevinek P et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: A phase 3, open-label extension study (PERSIST). Lancet Respir Med 2014; 2(11):902-10
Ronan NJ, Einarsson GG, Twomey M et al. CORK Study in Cystic Fibrosis: Sustained Improvements in Ultra-Low-Dose Chest CT Scores After CFTR Modulation With Ivacaftor. Chest [Internet]. 2018;153(2):339‑48. https://doi.org/10.1016/j.chest.2017.10.005
Wainwright CE, Elborn JS, Ramsey BW et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med [Internet] 2015 Jul 16; 373(3):220-31. http://www.nejm.org/doi/10.1056/NEJMoa1709846
Rowe SM, Daines C, Ringshausen FC et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med [Internet] 2017; NEJMoa1709847. http://www.nejm.org/doi/10.1056/NEJMoa1709847
Davies JC, Moskowitz SM, Brown C et al. VX-659-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del Alleles. N Engl J Med [Internet] 2018; NEJMoa1807119. http://www.nejm.org/doi/10.1056/NEJMoa1807119
Keating D, Marigowda G, Burr L et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med [Internet] 2018 Oct 25;379(17):1612-20. http://www.nejm.org/doi/10.1056/NEJMoa1807120
Kerem E, Konstan MW, De Boeck K et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med [Internet] 2014;2(7):539-47. http://dx.doi.org/10.1016/S2213-2600(14)70100-6
Plant BJ, Goss CH, Plant WD, Bell SC. Management of comorbidities in older patients with cystic fibrosis. Lancet Respir Med [Internet] 2013; 1(2):164-74. http://dx.doi.org/10.1016/S2213-2600(13)70025-0
Quittner AL, Abbott J, Georgiopoulos AM et al. International Committee on Mental Health in Cystic Fibrosis: Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus statements for screening and treating depression and anxiety. Thorax 2016;71(1):26-34
Ronan NJ, Elborn JS, Plant BJ. Current and emerging comorbidities in cystic fibrosis. Press Medicale [Internet] 2017; 46(6P2):e125-38. http://dx.doi.org/10.1016/j.lpm.2017.05.011
Abbott J, Bilton D. Adherence to Ivacaftor is suboptimal. J Cyst Fibros [Internet]. 2015;14(5):547-8. http://dx.doi.org/10.1016/j.jcf.2015.08.001
Cystic Fibrosis Foundation. 2017 PATIENT REGISTRY A NNUAL DATA REPORT [Internet] 2017. www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf
Frayman KB, Sawyer SM. Sexual and reproductive health in cystic fibrosis: A life-course perspective. Lancet Respir Med 2015; 3(1):70-86
McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD. Fertility in men with cystic fibrosis: An update on current surgical practices and outcomes. Chest 2000; 118(4):1059-62
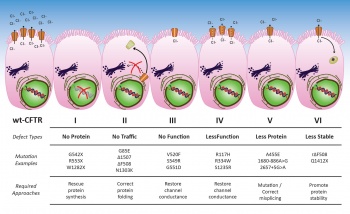 Figure 1: Overview for CFTR mutations and potential therapeutic approaches(click to enlarge)
Figure 1: Overview for CFTR mutations and potential therapeutic approaches(click to enlarge)