ENDOCRINOLOGY
Fibrous dysplasia: challenges in diagnosis and treatment
Fibrous dysplasia can be associated with several extra-skeletal manifestations that can complicate its diagnosis and treatment
February 3, 2016
-
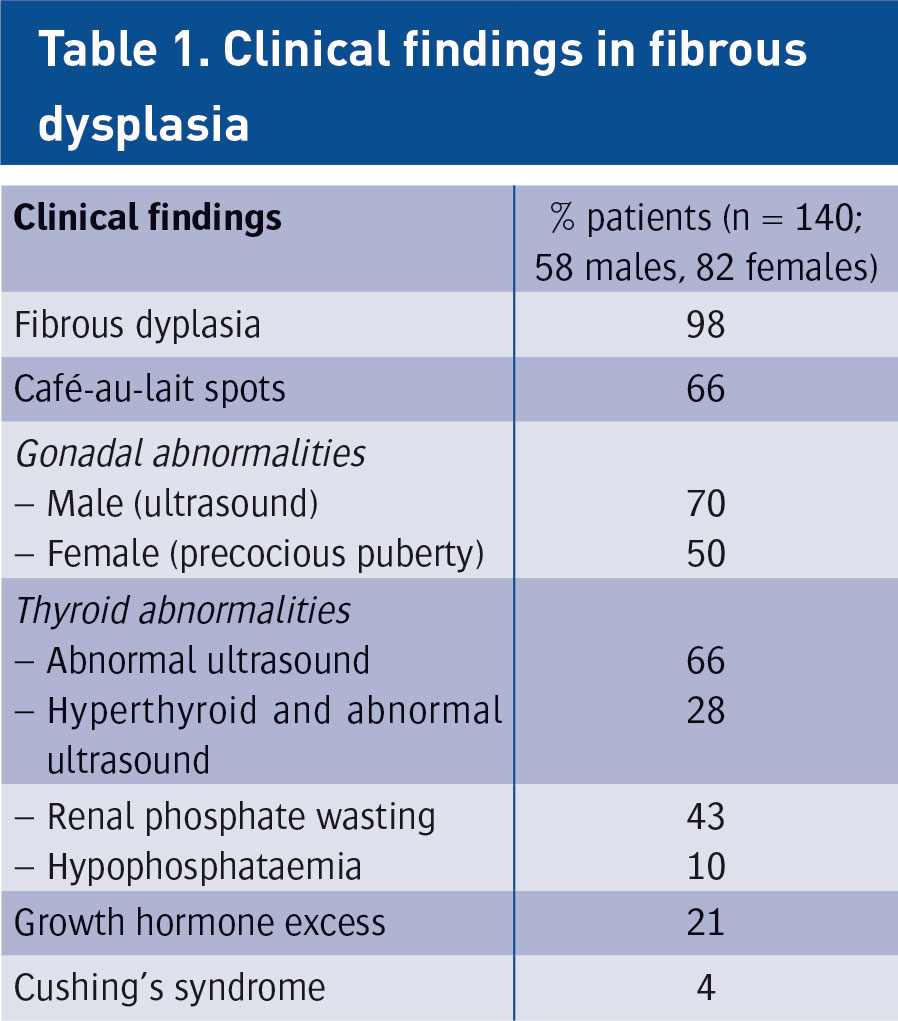
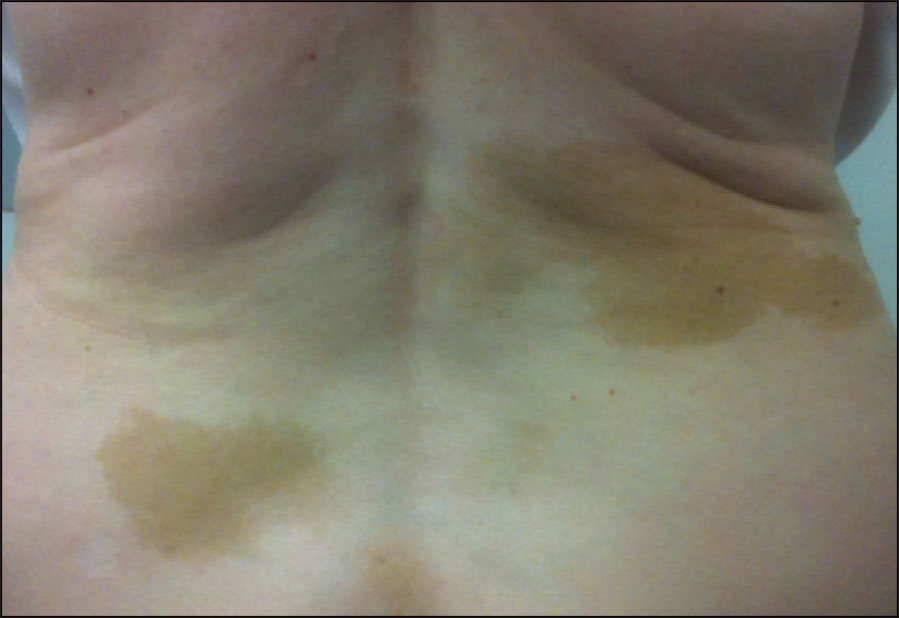
Fibrous dysplasia (FD) is an uncommon and debilitating skeletal disorder that typically presents to orthopaedic surgeons in Ireland for diagnosis and management. It can result in deformity, fracture, functional impairment and pain.1 Disease severity is highly variable and may affect one bone (monostotic FD) or many bones (polyostotic FD). Further complicating the diagnosis and treatment of FD is that it can be associated with a wide array of extra-skeletal manifestations. Common extra-skeletal manifestations include café-au-lait macules and hyperfunctioning endocrinopathies including hyperthyroidism, hypercortisolism, precocious puberty and growth hormone excess. One of the most common extra-skeletal manifestations of FD is a renal tubulopathy, which includes renal phosphate wasting. When extra-skeletal manifestations occur, the disease is referred to as McCune-Albright syndrome (MAS). The high degree of phenotypic variability makes the diagnosis and treatment of FD/MAS challenging.
Pathogenesis
FD arises from activating mutations in the GNAS gene, which encodes the alpha-subunit of the Gs stimulatory protein (Gs-alpha). Mutations arise postzygotically, leading to mosaic disease with a wide clinical variability. The stimulatory downstream effects of constitutively activated Gs-alpha results in increased adenylyl cyclase activity and inappropriate intracellular cyclic adenosine monophosphate (cAMP) production. Occurring in bone, this results in proliferation of undifferentiated bone marrow stromal cells resulting in marrow fibrosis and increased osteoclastogenesis. Bone trabeculae are abnormal in shape and biochemical composition, and in many cases are severely under-mineralised and abnormally compliant. Elevated serum levels of fibroblast growth factor 23 (FGF23), a phosphate-regulating hormone produced by highly activated osteoblastic cells in FD, are thought to result in the renal phosphate wasting associated with the extra-skeletal manifestations of FD. The pathological effects of Gs-alpha mutations in bone are more pronounced during phases of rapid bone growth, accounting for the fact that childhood and adolescence are when the disease most commonly presents.
Clinical features
The most common presenting features include pain, limp and fractures. Presentation before the age of five typically heralds more severe disease, likely to lead to greater morbidity. In general, children are less likely to complain of pain per se and may report stiffness or tiredness. In adults, presentation with pain is more common, especially in the long bones, ribs and craniofacial bones. Spinal and pelvic lesions are typically less painful. Pathological fractures or stress fractures in weight bearing limb bones are the main cause of morbidity. Deformities in bones are due to expansion and abnormal compliance of FD-affected bones and local complications such as cyst formation.
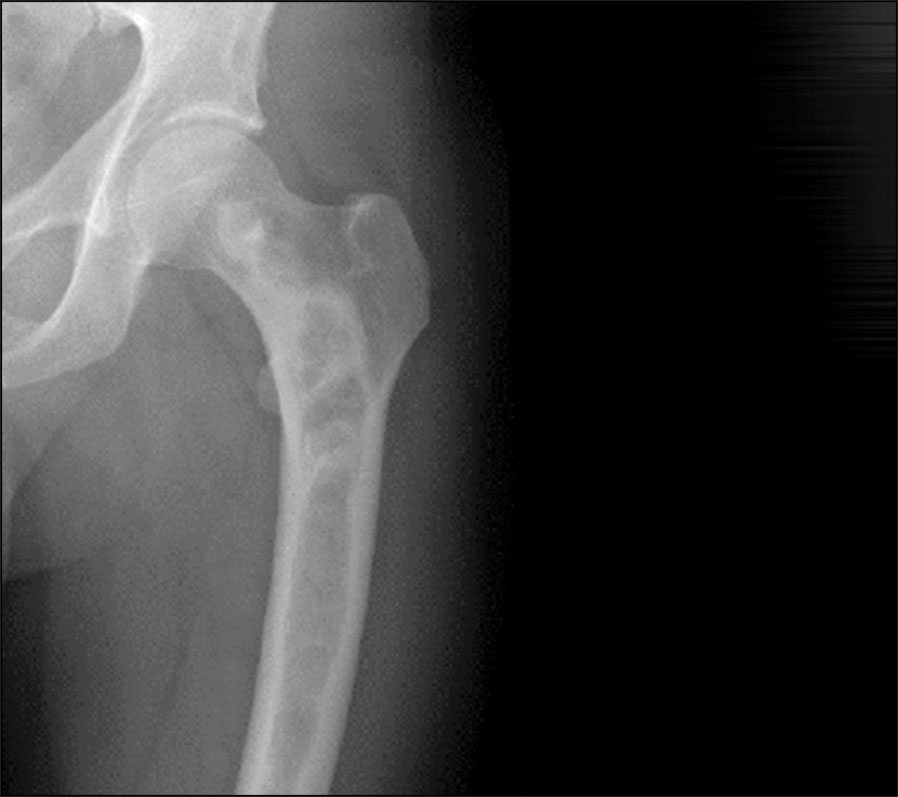
Diagnosis is usually made on clinical grounds, based on radiographic appearances and location. Areas of skeletal involvement are established early with 90% of craniofacial lesions evident by five years of age and 75% of all FD lesions established by 15 years of age. Although any area of the skeleton can be affected, common sites include the skull base and proximal femora. Skull base disease may result in facial asymmetry and, rarely, cranial nerve involvement. Femoral disease may present with a classic shepherd’s crook (coxa vara) deformity. Radiographically, these sites appear different. Craniofacial FD has a sclerotic appearance on plain film and ‘ground glass’ on CT; and over time they become more inhomogeneous and lytic on CT. Long bone disease typically displays a homogenous ‘ground glass’ appearance on plain film and becomes more sclerotic as the disease becomes quiescent with age (see Figure 1). The diagnosis can be supported by histological examination of biopsy material or mutation testing of the affected area. The majority of skeletal disease depending on the site is clinically manifest by three to 10 years of age. Markers of bone turnover are usually increased.
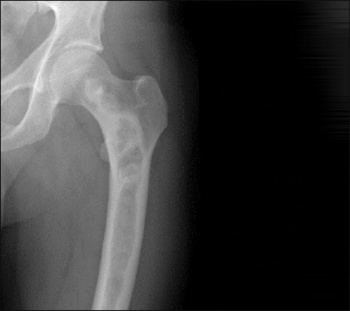 Figure 1. Radiographic findings on fibrous dysplasia in the femur(click to enlarge)
Figure 1. Radiographic findings on fibrous dysplasia in the femur(click to enlarge)