A framework for managing lipids and minimising risk
In asking how to manage high cholesterol, the answer will always be that it depends on a number of factors. Doctors will collaborate with patients to make the most informed decision
Dr Paddy Barrett, Consultant Cardiologist, Blackrock Clinic, Dublin
Any discussion on lipids generally comes down to one question: “Do I need to start a lipid-lowering therapy to lower my patient’s risk of a cardiovascular event?”
Lifestyle measures are recommended for all patients, and if a secondary cause is identified for elevated lipids that should be addressed first. The typical patient encounter arrives at the question: “My cholesterol is high – what should I do?” The answer, as always, is: “It depends”.
Although several guidelines are available to address this scenario, they can be cumbersome to read and, in general, most people look for the summary diagram and apply that.1
But as the saying goes: ‘Guidelines are like sausages. Everyone likes them, but no one wants to know what goes into them’. The truth is that when you read the guidelines in detail, they are generally incredibly informative. However, when you read the underlying studies, you realise the degrees of uncertainty that still exist in the literature.
Guidelines are also tilted towards providing recommendations on the evidence with the highest degree of validity, such as double-blind, randomised controlled trials (RCTs). While we should always prioritise such evidence, the clinical encounters we face are rarely represented by such evidence.
Most lipid-lowering RCTs range from two to five years in duration. But when we prescribe lipid-lowering therapy, we need to think about time horizons in the region of decades. To address this challenge, a better understanding of two key concepts is required:
What are lipids?
What causes atherosclerosis?
This article aims to provide an understanding of these concepts and a framework for clinical decision-making for patients with elevated lipids.
Lipids
Cholesterol does not traffic freely in plasma. It therefore does not come into direct contact with the arterial wall. Cholesterol is transported in a protein-based spherical structure, and the combination of these two parts makes up what is known as a lipoprotein. Each lipoprotein has a single Apolipoprotein B (ApoB) protein attached to it.2
The ApoB particle contacts the arterial wall and deposits the cholesterol into the subintimal space. The distinction needs to be made because the risk of atherosclerosis is directly related to the number of ApoB particles more so than the cholesterol concentration in these particles. Generally, these levels are linked on a 1:1 basis, so LDL (low-density lipoprotein) cholesterol concentration can be used as a surrogate marker for ApoB particle count, except when the patient has diabetes, metabolic syndrome or insulin resistance.2 The prevalence of these conditions is rising fast; therefore, the use of LDL cholesterol as the best risk marker is less than optimal. Ideally, an ApoB concentration should be measured to best assess risk, but in its absence, non-HDL (total cholesterol minus HDL-C) is sufficient.
Unfortunately, most guidelines, data models and clinical decision-making still rely on the use of LDL cholesterol. Because of this, we will use LDL-C as the marker of choice for discussion here, but we must always be mindful of its limitations.
What causes atherosclerosis?
Atherosclerosis is caused by the retention of an ApoB lipid particle in the subintimal space of the arterial wall. This retained particle then initiates an inflammatory cascade, ultimately resulting in the development of an atherosclerotic plaque.2
ApoB particles < 70nm in diameter passively flux across the endothelium to the subintimal space. This process can also occur actively in the presence of other risk factors such as diabetes, hypertension, smoking etc. This is why risk factors, in addition to high cholesterol, accelerate atherosclerosis.3
When an atherosclerotic plaque of sufficient size ruptures and causes a thrombus to form, this occludes the coronary artery and a myocardial infarction results.3
Atherosclerosis is the cause of myocardial infarction. The greater the burden of atherosclerosis, the greater the risk of myocardial infarction. The less the burden of atherosclerosis, the less risk.3 Our goal is to minimise the atherosclerosis burden for as long as possible for our patients.
Two factors now need to be considered:
Atherosclerosis begins to accumulate in arterial walls in the teenage years or even earlier4
By the age of 80, almost everyone will have a significant burden of advanced atherosclerosis.5
We all start accumulating atherosclerosis early in life and will accumulate a significant amount given a long enough time horizon. Some will accumulate atherosclerosis faster, some slower. But over a long enough period, essentially everyone will develop a clinically significant amount of atherosclerotic plaque. Some patients will die of non-cardiovascular causes before this arises.
This progressive accumulation of atherosclerosis is directly related to the lifetime exposure to ApoB lipid particles.3 The greater the exposure, the greater the plaque burden. Think of it like pack years of smoking, except as cholesterol years.
How much is too much?
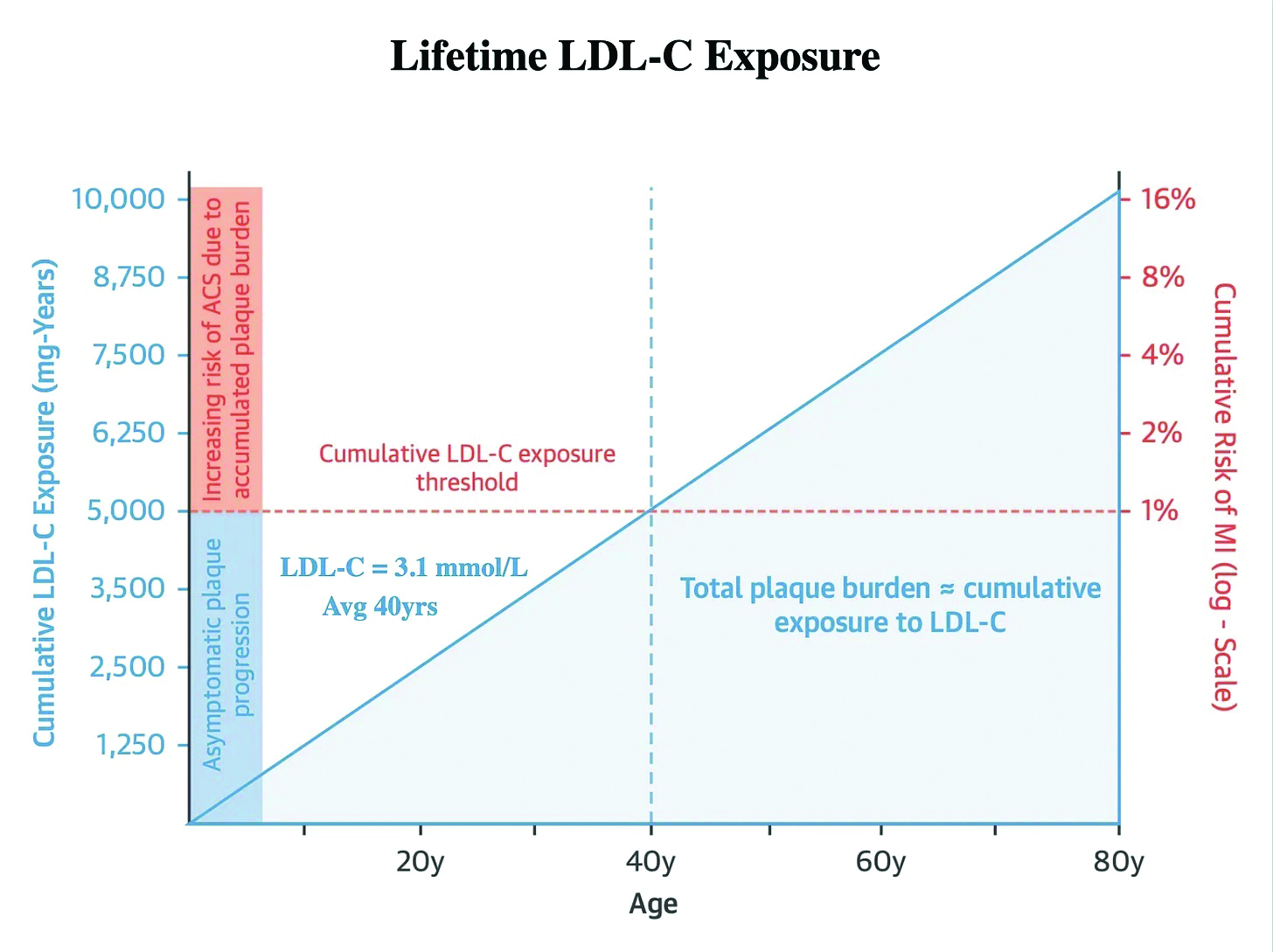
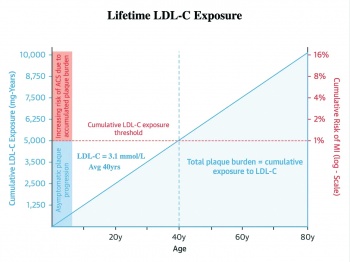
The cumulative incidence of myocardial infarction by the age of 40 is 1%. The average LDL cholesterol in adults is 3.1mmol/l. Therefore, 40 years of an LDL-C of 3.1mmol/l results in a sufficient burden of atherosclerosis to result in a 1% risk of myocardial infarction. The total cholesterol years then is approximately 124mmol/l years (3.1mmol/l x 40 years) (measured as 5,000mg/dl years in the US).3
Figure 1 illustrates this point graphically, with the blue shaded area representing the cumulative atherosclerotic plaque burden resulting in a 1% MI risk at age 40 that an average adult with a cumulative cholesterol of 124mmol/l years would have.3 The crucial point to appreciate is that the cumulative LDL-C concentration on the left of the graph rises linearly. In contrast, the risk of an event on the right side increases exponentially. This linear accumulation of LDL-C occurs with an LDL-C that stays at the same level. The only thing that is changing is time. However, the risk of an event doubles for each passing decade, with a 2% risk at age 50, a 4% risk at age 60 and an 8% risk at age 70. This is all in the setting of a stable untreated LDL-C of 3.1mmol/l.3
(click to enlarge)
10-year risk calculators
We must now consider how the same patient would be managed using a typical 10-year risk estimate, assuming they have relatively normal blood pressure and are non-smokers. Their 10-year risk is approximately 2%. Current guidelines would recommend lifestyle measures and to ‘consider’ drug therapy.
In reality, most patients in this category will not be treated with a lipid-lowering therapy. The most likely scenario is that they will be reassessed at age 50 or 60, where their risk based on a 10-year calculation will be more suggestive of commencing lipid-lowering therapy.
Based on the data provided, you can see the fallacy of such an approach. By the time the patient reaches 50 or 60, they will have accumulated a substantial additional burden of atherosclerosis and subsequent risk. Using risk alone as the marker to decide whether to initiate lipid-lowering therapy would only be reasonable if we had no idea what caused atherosclerosis. The cause of atherosclerosis is clear.
How to minimise risk
More atherosclerosis = more risk
Less atherosclerosis = less risk
Less atherosclerosis is a result of less cumulative lifetime LDL cholesterol exposure. Let’s see what happens when small changes are made earlier in life to result in less cumulative exposure by age 40.
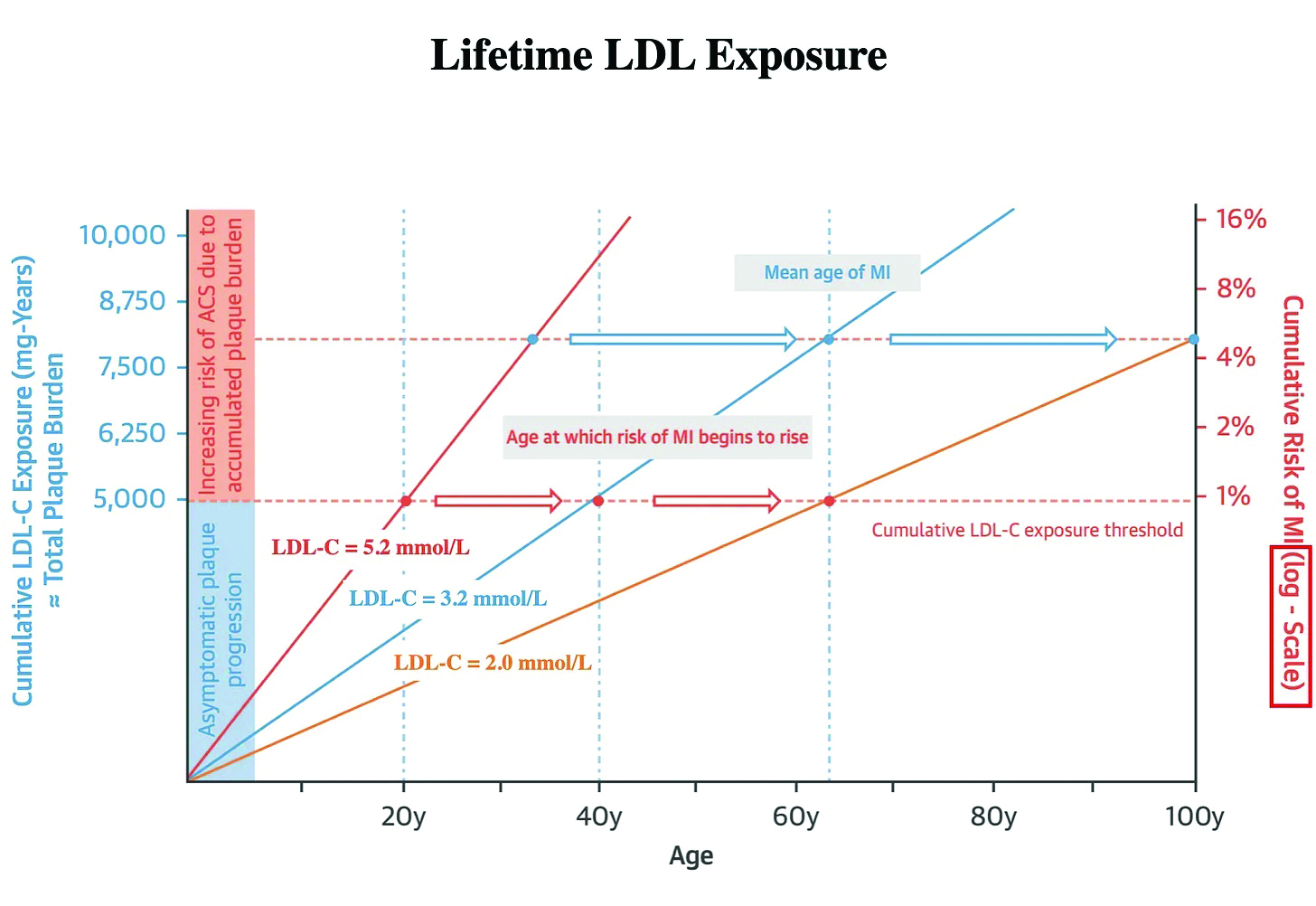
Figure 2 illustrates when the lifetime mean LDL cholesterol is 2mmol/l and 5.2mmol/l, respectively, at age 40.3 The mean age at which risk of MI begins to rise is approximately 64 years, which falls on the line representing a mean lifetime LDL-C of 3.2mmol/l. If this figure is reduced to an average lifetime LDL-C of 2mmol/l, the mean age at which risk of MI begins to rise is now pushed out to 100 years. If that average is increased to 5.2mmol/l, the mean age at which risk of MI begins to rise is projected to be in the 30s.3 This is why early, aggressive treatment of those with familial hyperlipidaemia is so important.
(click to enlarge)
The data used to generate these risk models are inferred from population-level data and not from RCTs. But how could they be when the duration of observation is multiple decades? Such trials will never be done. However, we can use Mendelian randomisation studies to reinforce that lifelong lower cumulative LDL cholesterol exposure substantially reduces cardiovascular risk. These natural RCTs assess those with gene variants that predispose individuals to either very low LDL-C or normal or high levels of LDL-C and evaluate their risk of a cardiovascular event over multiple decades.4 This is the scenario we are attempting to solve as clinicians.
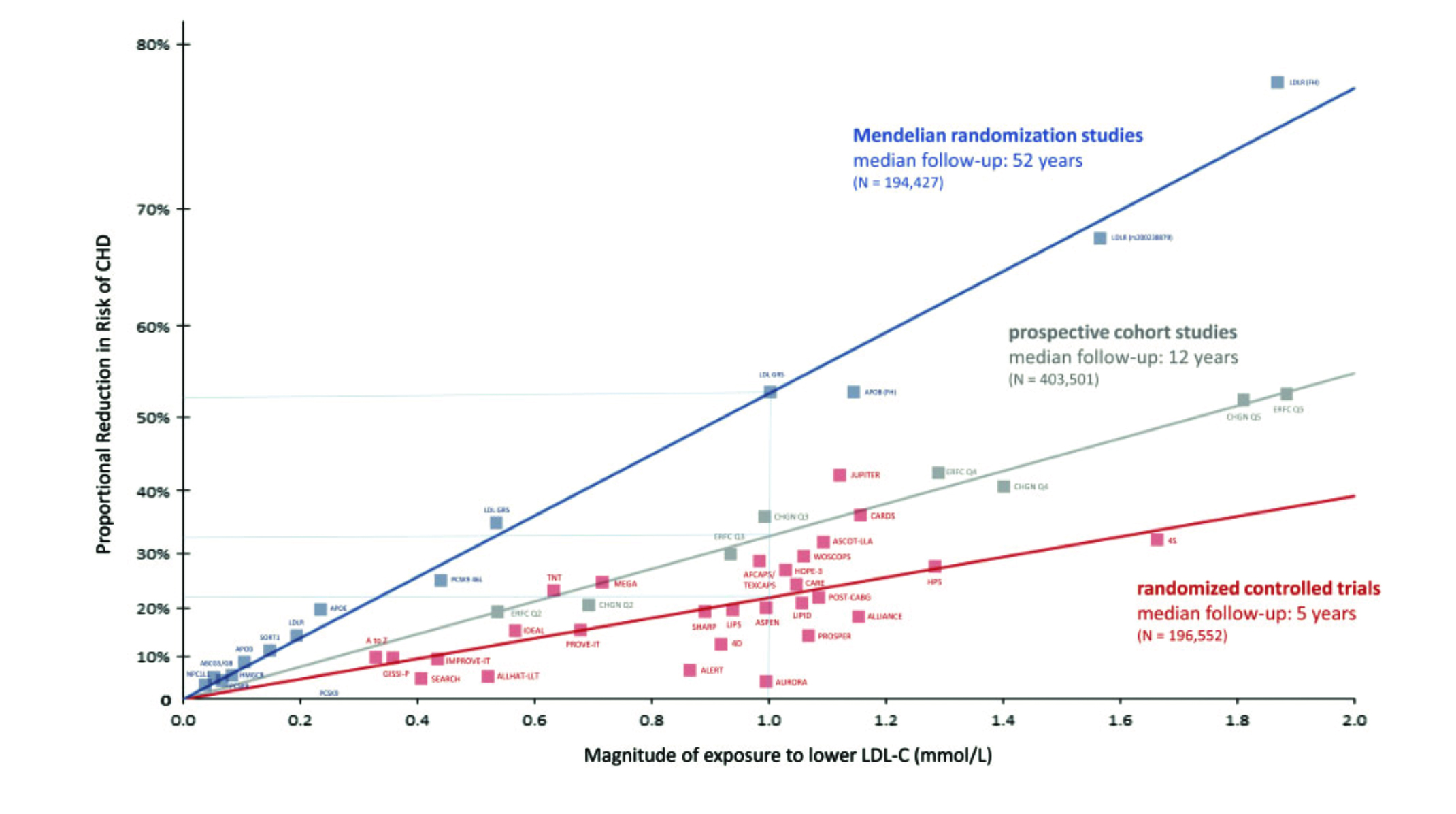
Figure 3 illustrates that those with lifelong low levels of LDL-C have reduced cardiovascular risk by up to 80%.6 Randomised trial data, however, show that lowering LDL cholesterol by the same amount only lowers this risk by 20-30%. The reason for the discrepancy is the time exposure to lowered LDL-C levels with the RCTs evaluating patients over an average of five years. In contrast, the Mendelian randomised trials assess patients for up to 50 years. This should reinforce the idea that risk relates to the cumulative exposure of LDL particles over time.
(click to enlarge)
How to lower LDL cholesterol
Approximately 50% of LDL cholesterol levels are influenced by genetics, and the other 50% by diet and lifestyle factors.7 Therefore, adhering to a diet that reduces LDL-C from early in life is an important step. Unfortunately, many people will be unable to sustain such lifestyle measures. Additionally, even with their best efforts, they may not be able to achieve an optimal level of LDL-C to reduce their level of lifetime risk.
Primary prevention relates to the management of risk factors once they have occurred. Primordial prevention relates to modifying the environments in which we live to prevent such risk factors even occurring. Optimal nutrition, exercise, work and home environments can influence whether we develop cardiovascular risk factors.
Members of the Tsimane tribe in Bolivia who have subsistence lifestyles have average LDL cholesterol of 2.35mmol/l and exceptionally low cardiovascular disease rates.3 However, most of us will not live in such environments and must navigate the challenges of modern life with easy access to processed foods etc. While it would be ideal if reliance on lifestyle factors alone were sufficient to achieve optimal lifetime LDL-C measures, it is unlikely to be possible for most.
Lipid-lowering therapies
When choosing a particular therapy to lower LDL cholesterol, what matters is the degree of lowering, not the drug choice. Historically, statin therapy was the sole option for lowering LDL-C and has repeatedly demonstrated reductions of LDL cholesterol in the order of 20-50%, with the expected reductions in cardiovascular events also.8
Each statin has differing characteristics. Do not get distracted by these. The objective is to lower the LDL-C in such a way that the patient tolerates it. Use whatever works. For most individuals, isolated statin therapy is all that will be required.
Statins work by increasing the expression of hepatic LDL receptors so that more LDL particles are removed from the circulation and are therefore not exposed to the arterial wall. Most of this effect is achieved in the low to moderate dose ranges, so the decision to add an alternative therapy such as ezetimibe should be made earlier in the titration pathway, rather than later. Use whatever gets the patient to target.
More recent lipid-lowering therapies include bempedoic acid,9 which has been shown to reduce LDL cholesterol concentrations by 25% when used in isolation and up to 40% when used in combination with ezetimibe.9
PCSK9 inhibitors are a well-established lipid-lowering option for those at very high risk of events. In isolation, LDL-C levels can be reduced by approximately 60% and up to 85% when combined with other oral therapies.10 Unfortunately, access to such treatments is highly restrictive.
Inclisiran is a novel twice-yearly small interfering RNA therapy that can reduce LDL-C levels by up to 60%.11 Phase III outcome trial data is awaited, but the convenience and degree of LDL particle lowering is impressive.3 Predictably, access to this therapy is likely to be a challenge.
Lastly, Verve Therapeutics recently announced the development of a PCSK9 gene-silencing technology that may function as an LDL cholesterol ‘vaccine’ with a single dose achieving lifelong large LDL cholesterol reductions.12
When prescribing a lipid-lowering therapy to my patients, they often ask: “Will I be on this for life?”. My answer is always the same. “No – only until something better comes along”. And that ‘better’ is on the horizon.
The choice of therapy is unlikely to be the major factor. What matters most is that the LDL cholesterol is adequately reduced.
What about LDL cholesterol targets?
When the decision is made to initiate lipid-lowering, the next question is, what target are you aiming for? Current guidelines suggest that the target LDL cholesterol is based on the patient’s risk:
For those at low risk, an LDL-C of < 3.0mmol/l is
recommended
For moderate risk < 2.6mmol/l
For high risk < 1.8mmol/l
For very high risk < 1.4mmol/l.
What is often missed in these recommendations is the suggestion to achieve a 50% LDL-C reduction and reach the pre-specified target.13 So for some individuals, the number targeted will be much lower than is attached to their assigned risk. But where do these targets come from? Again, it is always better to have a framework of understanding rather than a memory of numeric values. Again, more atherosclerosis equals more risk.
Less cumulative lifetime cholesterol exposure = less progression of atherosclerosis = less risk.
Therefore, lower is better. But why the figures of 1.8mmol/l and 1.4mmol/l?
Intravascular ultrasound studies done before and after aggressive lipid-lowering therapy routinely show that those who achieve LDL-C levels of < 1.8mmol and particularly < 1.4mmol LDL-C show plaque stabilisation and regression. This has been safely demonstrated down to levels of < 1.0mmol/l LDL-C. If atherosclerotic plaque is regressing at these levels, then there is less plaque and, therefore, less risk. The literature investigating these targets has demonstrated this.14
The 2021 ESC guidelines on cardiovascular disease prevention has one of my favourite messages: “Treatment recommendations are never ‘imperative’ for high-risk patients, nor are interventions ever ‘prohibited’ for patients at low to moderate risk.”1
When deciding how to manage a patient with high cholesterol, the answer will always be: “It depends”. It will depend on the patient’s age, cumulative cholesterol exposure, other risk factors, their goals, their concerns regarding medications, the time horizon over which they are looking to make a difference and their personal risk tolerance.
If the answer to when to lower LDL-C was as easy as “If LDL-C > X, then prescribe Y”, I don’t see why doctors should even be involved in the process. But that’s not what doctors do. We integrate all objective facts with the individual patient’s goals and concerns, and collaborate with them to make the most informed decision. We understand that we will only ever be dealing in probabilities, not certainties. We don’t have all the answers. But we do have some excellent clues.
References
ESC National Cardiac Societies; ESC Scientific Document Group. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J 2021 Sep 7; 42(34):3227-3337. doi: 10.1093/eurheartj/eha
Sniderman A et al. Update on apolipoprotein B. Curr Opin Lipidol 2021 Aug 1; 32(4):226-30. doi: 10.1097/MOL.0000000000000754
Ference BA et al. Impact of lipids on cardiovascular health: JACC Health Promotion Series. J Am Coll Cardiol 2018 Sep 4; 72(10):1141-56. doi: 10.1016/j.jacc.2018.06.046
Tuzcu EM et al. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: evidence from intravascular ultrasound. Circulation 2001 Jun 5;103(22):2705-10. doi: 10.1161/01.cir.103.22.2705
Tota-Maharaj R t al. Coronary artery calcium for the prediction of mortality in young adults <45 years old and elderly adults >75 years old. Eur Heart J 2012 Dec; 33(23):2955-62. doi: 10.1093/eurheartj/ehs230
Ference BA et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017 Aug 21; 38(32):2459-72. doi: 10.1093/eurheartj/ehx144
Zhang Y et al. Association between cumulative low-density lipoprotein cholesterol exposure during young adulthood and middle age and risk of cardiovascular events. JAMA Cardiol 2021;6(12):1406-13. doi: 10.1001/jamacardio.2021.3508
Coronary computed tomography angiography from clinical uses to emerging technologies: JACC State-of-the-Art Reviews. JACC 2020 Sep 8; 76(10):1226-43. doi: 10.1016/j.jacc.2020.06.076
CLEAR Harmony Trial. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N Engl J Med 2019 Mar 14; 380(11):1022-32. doi: 10.1056/NEJMoa1803917
FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med 2017 May 4; 376(18):1713-22. doi: 10.1056/NEJMoa1615664
ORION-10 and ORION-11 Investigators. Two Phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N Engl J Med 2020 Apr 16; 382(16):1507-19. doi: 10.1056/NEJMoa1912387
ESC Committee for Practice Guidelines (CPG); ESC National Cardiac Societies. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019 Nov; 290:140-205. doi: 10.1016/j.atherosclerosis.2019.08.014
Nicholls SJ et al. Effect of evolocumab on progression of coronary disease in statin-treated patients. The GLAGOV Randomized Clinical Trial. JAMA 2016, Dec 13; 316(22):2373-84. doi: 10.1001/jama.2016.16951
 (click to enlarge)
(click to enlarge)