Idiopathic pulmonary fibrosis: diagnosis and management
IPF is often well established by the time of diagnosis as patients frequently attribute their symptoms to advancing age
Ms Nicola Cassidy, Commitee, the Irish Lung Fibrosis Association, Dublin and Prof Jim Egan, Respiratory Physician, Mater Misericordiae University Hospital, Dublin
Idiopathic Pulmonary Fibrosis (IPF) is a chronic, progressive, irreversible and terminal fibrotic lung disease of unknown origin.1
It is the most common and severe form of the diffuse interstitial lung diseases (ILD). Life expectancy is seriously curtailed with IPF and the prognosis is worse than many malignancies.1
Epidemiology and disease progression
The estimated prevalence for IPF in Europe ranges from 1.25 to 23.4 cases per 100,000,2 with more older adults affected. However, robust epidemiological data are lacking. In Ireland, the prevalence is estimated at 800-1,000 individuals based on extrapolations from data for the UK where the prevalence is 15,000 cases.3 The incidence of IPF is rising in Europe and an estimated 40,000 new cases are diagnosed annually.3 The median age for diagnosis is 60-70 years and IPF predominates in males. Median survival time is two to three years following diagnosis, however, the natural course of disease progression is highly variable and unpredictable.
Some patients deteriorate slowly, some decline rapidly, a minority remain stable for years, and others experience acute exacerbations, which are associated with high mortality.
An acute exacerbation is an unexplained rapid deterioration of the patient’s clinical status with progression of fibrosis with new pulmonary infiltrates and respiratory decline in the absence of infection, pneumothorax, congestive heart failure or other known causes.1,4
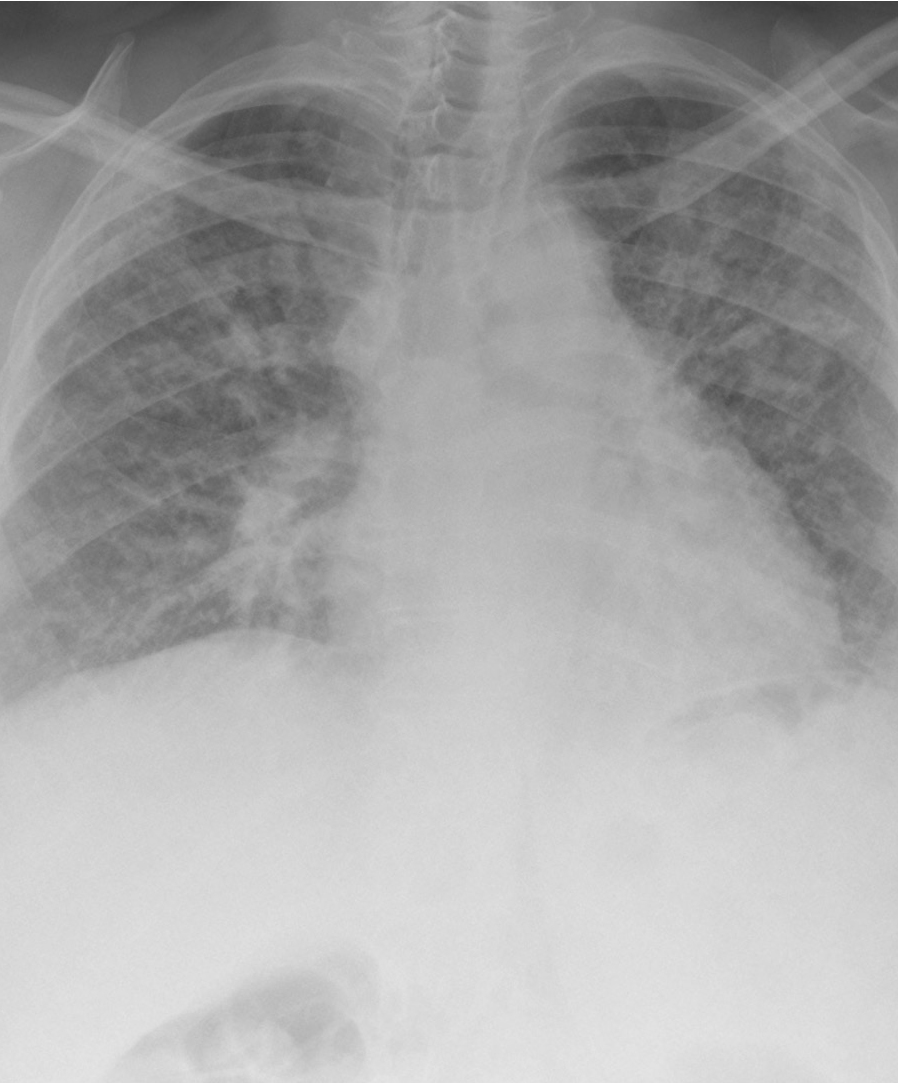
Severe pulmonary fibrosis(click to enlarge)
Pathophysiology
The underlying pathology of IPF consists of repetitive epithelial injury and abnormal fibrogenesis with minimal or no inflammation. Aberrant lung fibrosis causes an irreversible extracellular matrix remodelling with destruction of the parenchymal architecture.5
IPF results in restricted lung volume, progressive impairment of gas exchange parameters and hypoxaemia. Patients ultimately die from respiratory failure secondary to worsening lung function and progressive fibrosis, or from other disease-related complications.1
Contributory factors such as smoking or a history of previous smoking, environmental exposures (eg. infection, mould, viruses, bird droppings), occupational exposures to dusts and chemicals, collagen vascular disease and drug toxicity are believed to confer an increased risk for developing an ILD in susceptible individuals. However, the causative agent(s) and trigger(s) that are critical to IPF development are unknown.6,7
Familial forms of IPF exist, but account for less than 5% of total cases. There is no distinguishable difference between familiar and sporadic forms, although familial IPF may be seen in younger patients. Genetic testing is not currently recommended.1
Diagnosis
IPF is a complex diagnosis of exclusion using a combination of evidence-based clinical, radiological and pathological findings to exclude other known causes of ILD. Misdiagnosis is common and is complicated by comorbidities including emphysema (secondary to smoking) and bronchiectasis. Patients usually present with a history of gradual worsening dyspnoea on exertion, fatigue and a non-productive cough, all of which impact significantly on their quality of life. Finger-clubbing may be present and is more common in males.
A key non-specific clinical feature is the sound of basal inspiratory crackles on lung auscultation, often referred to as ‘velcro’ crackles; the sound made when velcro is torn apart. Audio samples of velcro-crackles can be listened to on www.soundsofipf.com8
A chest X-ray is usually non-specific and cannot accurately diagnose IPF, particularly in the early stages of disease. A high resolution computed tomography (HRCT) thoracic scan is warranted to categorise the underlying lung aetiology. Honeycombing, a diagnostic and distinctive fibrosis pattern on HRCT that shows sub-plural basal-predominant reticular abnormalities, is indicative of advanced IPF. This pattern can be difficult to accurately identify even for experienced radiologists.
A surgical lung biopsy may be required for a minority of patients to help definitively diagnose IPF, however risk factors including advanced age or comorbidities may preclude patients from this invasive and non-essential procedure.1
IPF is often well established by the time of diagnosis as patients frequently attribute their symptoms to advancing age and may not seek timely medical advice. Indeed, clinicians may initially overlook the patient’s symptoms on account of a previous smoking history. Early referral to a specialist centre is paramount if an ILD is suspected, to ensure a more accurate diagnosis and initiation of appropriate treatment.9
A higher mortality rate is associated with a longer delay from the onset of dyspnoea to referral to a specialist centre, irrespective of disease severity10 and the diagnostic accuracy for IPF is superior with collaborative multidisciplinary input from professionals experienced in ILD.1
The Irish Thoracic Society published Guidelines on the Management of IPF in 2012 and recommended that patients be assessed in a designated IPF centre with experienced respiratory physicians, radiologists and pathologists with specialist expertise, a nurse specialist and rehabilitation services.11
In Ireland specialist IPF centres are located in the Mater Misericordiae University Hospital and St Vincent’s University Hospital in Dublin and in Cork University Hospital, Limerick University Hospital and Galway University Hospital.
IPF related complications
Comorbidities are common and gastroesophageal reflux disease (GERD), pulmonary hypertension, vascular or coronary artery disease, obesity, diabetes, emphysema and sleep apnoea are the most frequent concomitant conditions.1
GERD is extremely common and results in a vicious cycle of recurrent epithelial injury secondary to micro-aspiration of gastric fluid, wound repair and fibrosis. Some 33-50% of IPF patients with GERD are asymptomatic. Pulmonary hypertension is common and is associated with advanced disease and a worse prognosis.1 These patients are also at increased risk of developing an acute exacerbation and increased mortality.4
Management
The irreversible nature of the fibrosis effectively rules out a pharmacological cure. Consequently, current treatment modalities are directed towards:
Slowing disease progression
Supportive care and treating disease-related complications
Palliative care to alleviate symptoms and improve quality of life
Lung transplantation.
An orderly approach with a combination of pharmacological and non-pharmacological interventions is needed. Regular clinical evaluations are required due to the progressive nature of IPF and physicians should initiate timely advanced care planning in partnership with the patient and family members.
Non-pharmacological treatments
Supplemental oxygen is crucial for the management of IPF and is indicated when patients have significant resting hypoxaemia (defined as a resting SpO2 <88%). Patients may require 24-hour high-dose oxygen as their lung function and hypoxaemia progressively deteriorate.1 Pulmonary rehabilitation, a structured programme that focuses on education, exercise and maximising functional ability, has proven benefits for IPF patients and improves six-minutes walk distance, dyspnoea and fatigue.12
Pulmonary rehabilitation courses are restricted to a limited number of hospitals and community facilities in Ireland, but are not widely accessible for IPF patients.
Pharmacological treatments
Pirfenidone is the only licensed pharmacological agent approved for the treatment of mild to moderate IPF (defined as forced vital capacity predicted <50%). Pirfenidone inhibits the expression of certain growth factors and release of pro-inflammatory cytokines. Studies have shown clinically meaningful results for improved lung function, disease-free progression survival, and mortality for patients with mild to moderate IPF.13 Pirfenidone was approved in Ireland in 2013 for prescribing by IPF specialists.
Nintedanib is a triple tyrosine kinase inhibitor and a potent antagonist of a number of growth factors involved in signalling pathways that lead to angiogenesis and fibrosis. In clinical trials, nintedanib significantly reduced the rate of decline in forced vital capacity (FVC) at one year and sowed the progression of IPF in patients with mild to moderate IPF.14 Nintedanib was licensed by the FDA recently for the treatment of IPF and was validated and granted accelerated assessment by the European Medicines Agency in June this year.
Lung transplantation
Lung transplantation is the only effective treatment for prolonging life in IPF in selected patients. Approximately 30% of international lung transplantations occur in IPF patients15 and in Ireland the five-year survival is exceptionally high at 91%.16 Early referral for lung transplantation assessment is essential in order to identify suitable candidates. IPF patients awaiting lung transplantation have higher mortality than patients with other respiratory conditions.16
In 2013, 32 lung transplantations were performed at the National Lung Transplant Unit at Mater Misericordiae University Hospital and 12 recipients had IPF.
Patient support
Patients should be encouraged to take a proactive approach to their health and engage with their healthcare team to optimise the management of their IPF. Psychosocial support through counselling and patient support groups can play a vital role in allowing patients to set goals, become more functional and attain a better quality of life.
The Irish Lung Fibrosis Association (ILFA) is a voluntary charity founded in 2002 to support patients and families living with lung fibrosis and to raise awareness about IPF. ILFA is acknowledged as one of the leading IPF advocacy charities in Europe. ILFA’s positive ethos concentrates on quality of life, exercise, and focuses on what patients can achieve despite their limitations.
ILFA’s educational resources are developed specifically for IPF patients and are peer reviewed by patients to ensure they are appropriate. ILFA holds two patient information days annually and four patient support groups meet monthly in Dublin, Cork, Kerry and Offaly.
In 2013, ILFA developed a free home-based exercise programme for IPF patients called ‘The ILFA 2000 Steps a Day Challenge’ for IPF patients to encourage exercise and wellbeing. For more information see www.ilfa.ie
References
Raghu G, Collard HR, Egan JJ et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183(6):788-824
Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012;21(126):355-36
Navaratnam V, Fleming KM, West J et al. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011;66:462-467
Judge EP, Fabre A, Adamali HI, Egan JJ. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Respir J 2012; 40: 93–100
Selman M, King TE, Parddo A. Idiopathic Pulmonary Fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001:134,136-151
Spagnolo P, Tonelli R, Cocconcelli E, Stefani A, Richeldi L. Idiopathic pulmonary fibrosis: diagnostic pitfalls and therapeutic challenges. Multidisciplinary Respiratory Medicine 2012;7:42
Molyneaux PL and Maher TM. The role of infection in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir Rev 2013:22;376-381
www.soundsofIPF
Cottin V. Interstitial lung disease. Eur Respir Rev 2013:22:26-32
Lamas DJ, Kawat SM, Bagiella E, Philip N, Arcosoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis. Am J Resp Crit Care Med 2011:184;842-847
Position Statement from the Irish Thoracic Society on the treatment of Idiopathic Pulmonary Fibrosis. http://www.irishthoracicsociety.ie
Swigris DJ, Fairclough, DL, Morrison M, Make B, Kozora E, Brown KK, Wamboldt FS. Benefits of pulmonary rehabilitation in Idiopathic Pulmonary Fibrosis. Respir Care 2011;56(6):783-789
King TE Jr, Bradford WZ, Castro-Bernardini S et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014:29;370(22):2083-92
Richeldi L, du Bois RM, Raghu et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014:29;370(22):2071-2082
Christie JD, Edwards LB, Kucheryavaya AY et al. The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart-lung transplant report 2012. J Heart Lung Transplant 2012:31(10);1073-86
Adamali HI, Judge EP, Healy D et al. International collaboration: a retrospective study examining the survival of Irish citizens following lung transplantation in both the UK and Ireland. BMJ Open 2012:28:2(2):e000605 doi:10.1136/bmjopen-2011-000605
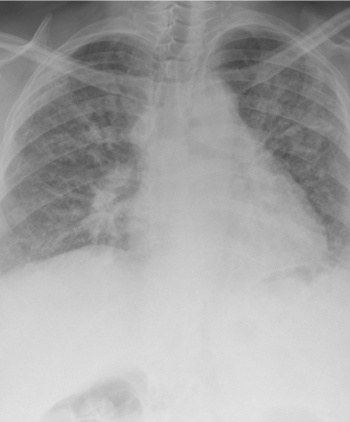 Severe pulmonary fibrosis(click to enlarge)
Severe pulmonary fibrosis(click to enlarge)