Unexplained long bone fractures and soft tissue sarcomas
A case of neurofibromatosis type 1 (NF-1) in an eight-month-old female infant who had pseudoarthrosis of tibia and fibula, café-au-lait spots and a soft tissue sarcoma
Dr Muhammad Jawad, Locum Consultant Paediatrician, Wexford General Hospital, Wexford
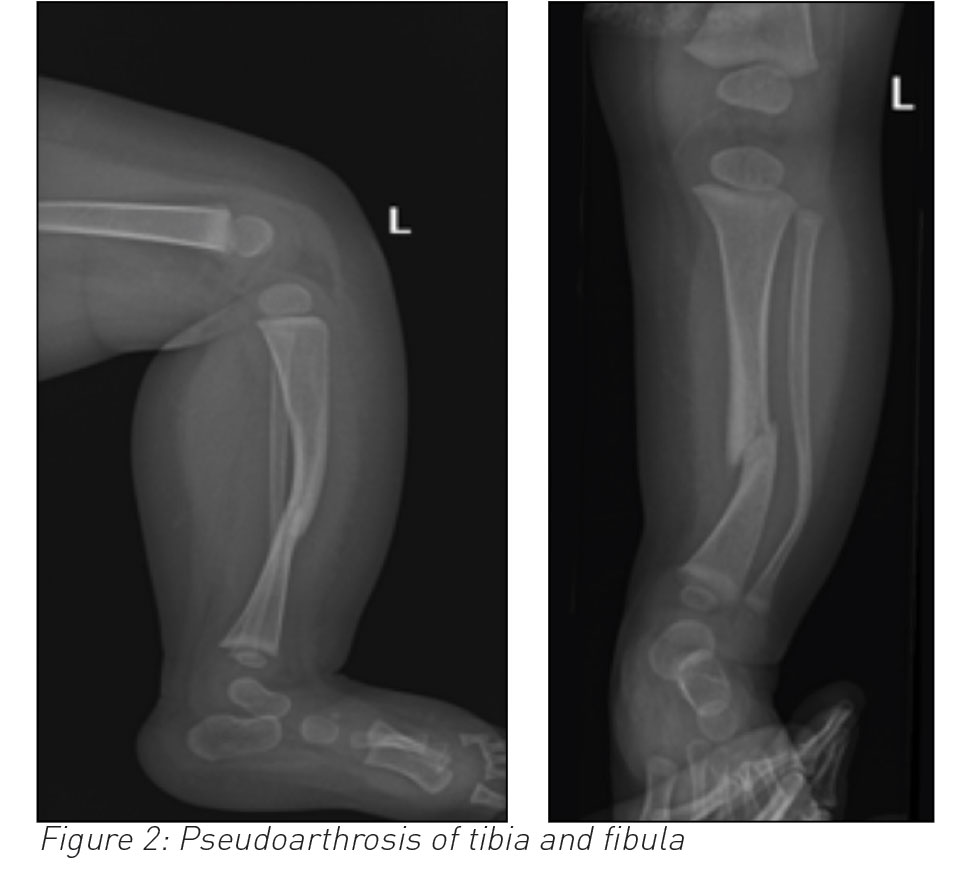
An eight-month-old non-mobile female infant presented with a history of not moving her left leg for two days and displayed discomfort on passive movement of her left foot. She underwent an x-ray of her left leg, revealing fracture of both tibia and fibula along with antero-lateral bowing. It was strongly considered for her to have a non-accidental injury (NAI). She underwent further tests to investigate the non-accidental injury and had her fracture splinted.
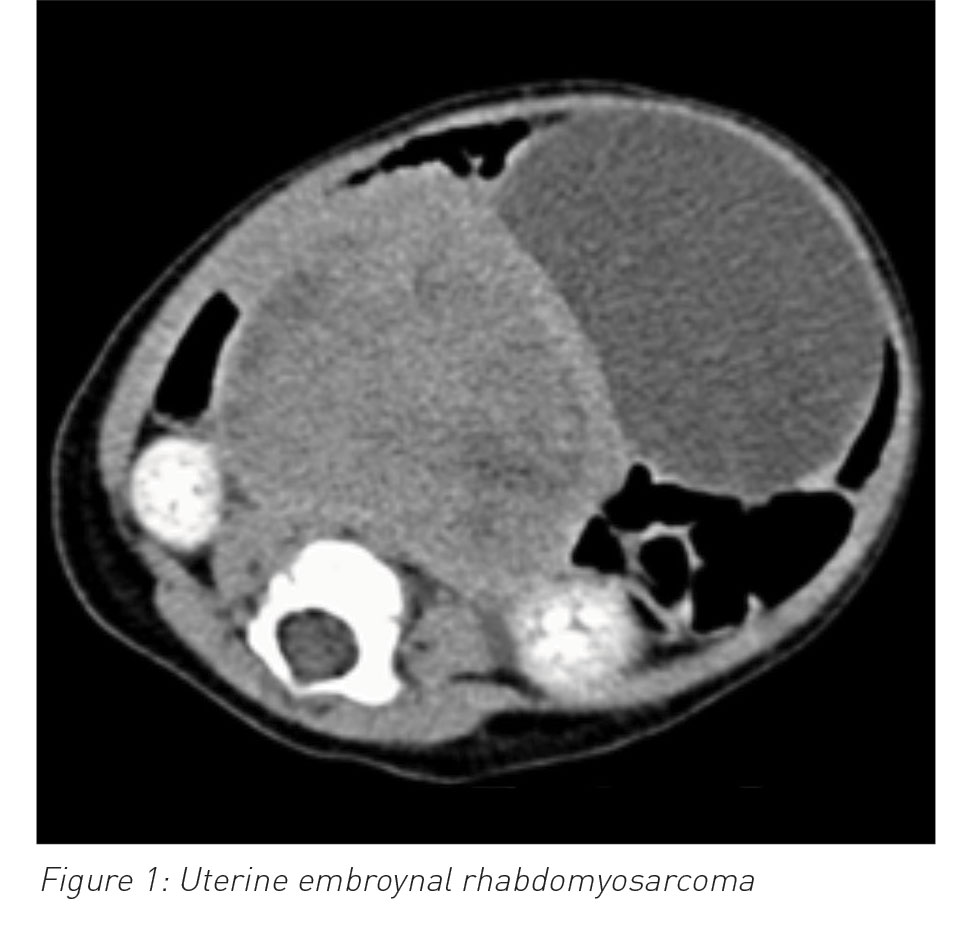
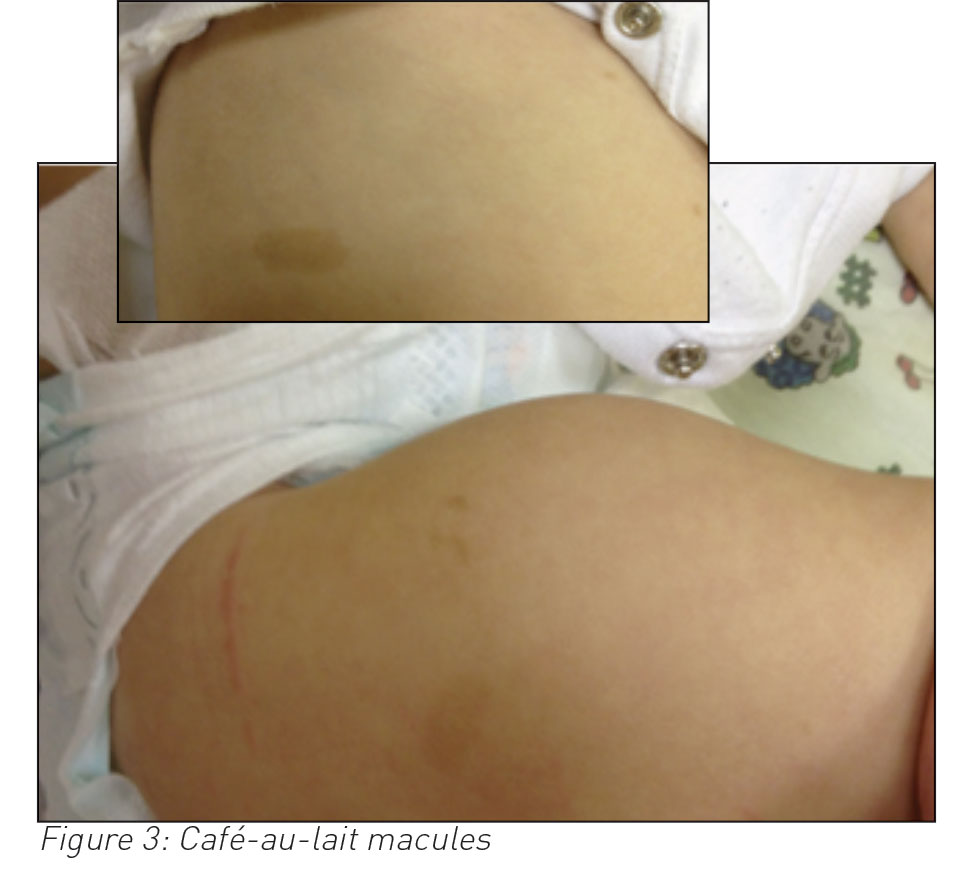
She presented again two weeks later with irritability, lower abdominal swelling and vaginal bleeding. A few small faint macules noted at her first presentation had turned brownish in colour on her thighs and abdomen. She had an ultrasound and CT scan of her abdomen and pelvis, which showed a uterine mass with no metastasis in the abdominal or pelvic cavity. Uterine tumour histology was compatible with a diagnosis of embryonal rhabdomyosarcoma (ERMS).
The infant was born premature but had normal development and was thriving well.
She was diagnosed by a clinical geneticist a case of neurofibromatosis type 1 (NF-1) based on the clinical criteria of pseudoarthrosis of tibia and fibula, café-au-lait spots and a soft tissue sarcoma. There was no family history of NF-1. She was likely to be a case of de novo mutation.
She had a hysterectomy followed by chemotherapy and radiotherapy afterwards. She responded well to the above treatment and was making good clinical progress.
(click to enlarge)
Discussion
Neurofibromatosis is an autosomal dominant neurocutaneous condition caused by mutation in NF-1 gene. The complications are diverse and disease expression varies among families.1
The incidence is approximately one in 3,000 individuals.1 Approximately half of NF-1 cases are familial (inherited); the remainder represent sporadic mutation, as in our case.
The diagnosis is mainly clinically based on presence of two or more cardinal features as shown below. Genetic testing is not usually required to make a diagnosis but is helpful in cases not meeting the clinical diagnostic criteria.
Diagnostic criteria for neurofibromatosis type 1
In order to make a diagnosis of NF-1, two or more of the following clinical features must be present:
Six or more café-au-lait macules of more than 5mm in greatest diameter in prepubertal individuals, and more than 15mm in greatest diameter in postpubertal individuals
Two or more neurofibromas of any type or one plexiform neurofibroma
Freckling in the axillary or inguinal regions
Optic glioma
Two or more iris hamartoma (Lisch nodules)
Distinctive bony lesion, such as sphenoid dysplasia, or thinning of the long bone cortex with or without pseudoarthrosis
A first-degree relative (parent, sibling or offspring) with NF-1 based on the above criteria.
Long bone dysplasia, almost always unilateral occurs in 3-5% of individuals with NF-1.2 The tibia is the most commonly affected long bone, often in association with the fibula. Once the long bone fractures, non-union (pseudoarthrosis) frequently occurs. The shortening of the affected limb occurs in younger patients because of tibial bowing and poor growth in distal tibial epiphesis.3,4
Pseudoarthrosis of the tibia and fibula
Treatment of pseudoarthrosis of long bone is still a challenge. Each treatment aims to obtain a long-term bony union of dysplastic fracture site to prevent limb length discrepancy, to avoid soft tissue lesions and associated joint stiffness. An invasive method of treatment comprises stable internal or external fixation of pseudoarthrotic site with a bone graft. A noninvasive method, which includes electrical stimulation without surgery, is also described to enhance union. However, neither has yet proven its superiority.4
The risk of malignancies in NF-1 patients is estimated to be 5-15% higher than in the general population.5 The tumours generally occur at an early age and often have a neural crest and myeloid origin.5 The common tumours are neurofibromas which are benign but malignant peripheral nerve sheath tumours, glioma, leukaemia and brain tumours are also seen.6
Rhabdomyosarcomas are encountered 20 times more frequently in patients with NF-1 than in the general population. They tend to present at an early age and often arise in a genitourinary site.
Often the diagnosis is made when the tumour is well advanced, but these tumours have generally a good response to chemotherapy.7
(click to enlarge)
(click to enlarge)
Conclusion
This case report highlights the importance of considering this neurocutaneous syndrome in the differential when encountering children with unexplained long bone fractures and soft tissue sarcomas.
References
Ferner RE, Huson SM, Kirby A. Guidelines for the diagnosis and management of individuals with NF 1. J Med Genet 2006 Feb; 44(2):81-88
Stevenson DA, Little D, Armstrong L et al. Approaches to treating NF1 tibial pseudoarthrosis: Consensus from the children’s tumor foundation NF1 bone abnormalities consortium. J Pediatrics Orthopedics 2013 April; 33(3):269-275
Stammers K, Cope S. Congenital pseudoarthrosis in a child with neurofibromatosis. BMJ Case Rep 2015. Doi: 10.1136/bcr-2015-212813
Shah H, Rousset M, Canavese F, Congenital pseudoarthrosis of the tibia: management and complications. Indian J Orthop 2012; 46:616-26
Scalzone M, Coccia P, Ruggiero A et al. The neurofibromatosis type 1: A dominantly inherited tumor predisposing disorder. Central European J of Med 2009 March 4(1):11-16
Hadjistilianou T, Masterangelo D, Gragnoli A et al. Letter to the editor: Neurofibromatosis type 1 (NF1) associated with embroynal rhabdomyosarcoma of the orbit. Med Pediatr Oncol 2002; 38:449
Korf BR. Malignancy in neurofibromatosis type 1. The Oncologist 2000 Dec; 5(6):477-85
 (click to enlarge)
(click to enlarge)